





























Genome-wide DNA methylation profiling using Infinium® assay
Abstract
Aims: Bisulfite sequence analysis of individual CpG sites within genomic DNA is a powerful approach for methylation analysis in the genome. The major limitation of bisulfite-based methods is parallelization. Both array and next-generation sequencing technology are capable of addressing this bottleneck. In this report, we describe the application of Infinium® genotyping technology to analyze bisulfite-converted DNA to simultaneously query the methylation state of over 27,000 CpG sites from promoters of consensus coding sequences (CCDS) genes. Materials & methods: We adapted the Infinium genotyping assay to readout an array of over 27,000 pairs of CpG methylation-specific query probes complementary to bisulfite-converted DNA. Two probes were designed to each CpG site: a ‘methylated’ and an ‘unmethylated’ query probe. The probe design assumed that all underlying CpG sites were ‘in phase’ with the queried CpG site due to their close proximity. Bisulfite conversion was performed with a modified version of the Zymo EZ DNA Methylation™ kit. Results: We applied this technology to measuring methylation levels across a panel of 14 different human tissues, four Coriell cell lines and six cancer cell lines. We observed that CpG sites within CpG islands (CGIs) were largely unmethylated across all tissues (∼80% sites unmethylated, β < 0.2), whereas CpG sites in non-CGIs were moderately to highly methylated (only ∼12% sites unmethylated, β < 0.2). Within CGIs, only approximately 3–6% of the loci were highly methylated; in contrast, outside of CGIs approximately 25–40% of loci were highly methylated. Moreover, tissue-specific methylation (variation in methylation across tissues) was much more prevalent in non-CGIs than within CGIs. Conclusion: Our results demonstrate a genome-wide scalable array-based methylation readout platform that is both highly reproducible and quantitative. In the near future, this platform should enable the analysis of hundreds of thousands to millions of CpG sites per sample.
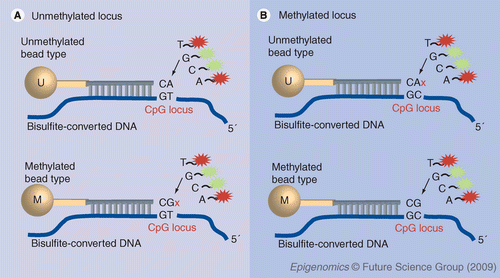
Nonmethylated cytosines (C) are converted to uracil (U) when treated with bisulfite, while methylated cytosines remain unchanged. Genomic DNA is bisulfite converted and whole-genome amplified using Infinium protocol. We make an assumption that adjacent CpG sites tend to be co-methylated or co-demethylated. Each CpG locus is represented by two bead types. One bead type (U) presents a probe that is designed to match to the unmethylated site; the second bead type (M) matches the methylated state. (A) On the left side of this figure, the locus of interest is unmethylated. It matches perfectly with U probe, enabling single-base extension and detection. The unmethylated locus has a single-base mismatch to the M probe, inhibiting extension that results in low signal on the array. (B) If the CpG locus of interest is methylated, the reverse occurs: the M bead type will display a signal, and the bead type will show a low signal on the array. If the locus has an intermediate methylation state, both probes will match the target site and will be extended. Methylation status of the CpG site is determined by the β-value calculation, which is the ratio of the fluorescent signals from the methylated probe to the total locus intensity.

Unmethylated (U), hemi-methylated (H), and methylated (M) reference standards were created from Coriell genomic DNA samples. Unmethylated gDNA was created by 100-fold whole-genome amplification of gDNA NA06999 and NA10924. The NA10924 amplified sample was treated with mung bean nuclease to remove single-stranded DNA, and then methylated with SssI methylase (M). The hemi-methylated reference was created by mixing U (NA06999) and M (NA10924) in a 1:1 ratio, and validated by genotyping.

(A) This plot shows reproducibility between technical replicates of bisulfite-converted Coriell DNA sample NA10923. (B) This plot shows correlation between fully converted NA10923 and the same sample spiked with 5% of unconverted DNA prior to the Infinium whole-genome amplification. Approximately 3.2% of CpG loci exhibit various shifts in β-values, resulting in a significant number of false-positive ‘differentially methylated’ loci.

(A) Correlation between technical replicates (starting from whole-genome amplification) in Infinium Methylation assay. (B) Sensitivity in detecting differential methylation depends on the β-value. Six DNA samples from various cell lines were analyzed in replicate on a 27k BeadChip. The average R2 was 0.986. From the noise in the replicates, the detectable Δβ is computed across the range of β-values. On average, a Δβ of 0.136 or larger is detectable with 95% confidence.

(A) This plot shows the correlation between Infinium array and bisulfite sequencing data for four CpG sites in the CD44 gene in six different DNA samples (20 clones per data point) (B) This plot Illustrates the correlation of β-values between Infinium and GoldenGate methylation assays across 113 different CpG sites in a Jurkat DNA sample.

Distribution of methylation levels in 14 normal tissues (A) and six cancer cell lines (B) in CpG sites located in (20,006) and outside (7572) of CpG islands.
In the recent years, the Human Epigenome Project (HEP) was initiated with one of the major goals to identify, catalogue and interpret genome-wide DNA methylation patterns of all human genes in all major tissues [101]. The success of this project depends on the development of novel strategies to analyze DNA methylation state across the human genome and generate detailed maps of the DNA methylome.
Changes in patterns of the cytosine methylation play a critical role in the regulation of gene expression [1,2], and may play an important role in cell fate specifications. Methylation in the human genome is generally limited to 5-methyl cytosine in the context of CpG sites. Various techniques for DNA methylation profiling were developed in the last two decades. These methods can be broadly divided into three main categories based on how the methylation status is interrogated: discrimination of bisulfite-induced C to T transition [3–9]; cleavage of genomic DNA by methylation-sensitive restriction enzymes [10–14]; and immunoprecipitation with methyl-binding protein or antibodies against methylated cytosines [15,16].
Each of these applications has its limitations. Methylation-sensitive restriction enzymes do not allow random access to specific sequences and cannot interrogate every CpG site; however, approximately a third of all CpGs in the genome can be assayed using a combination of enzymes [13] and, in combination with a high-density array readout can provide a powerful approach for genome-wide methylation profiling. The immunoprecipitation method overcomes the sequence-dependent limitation of all restriction digestion-based approaches, but cannot provide methylation information at single-base resolution for any targeted sequence. The challenges for the bisulfite-based approach lies in dealing effectively with the reduced genome complexity of the bisulfite-converted genome. The main remaining technical hurdles are the target-specific probe selection and hybridization specificity, which can be improved by incorporation of an enzymatic discrimination step, such as oligo ligation [17] and allele-specific extension [18], thus allowing multiplexed profiling of CpG methylation status in several hundred genes. Recent technical advances in array and genotyping technology are leading to development of more comprehensive, high-resolution genome-wide methods for epigenomic analysis [19–22]. Microarray-based DNA methylation profiling technologies have been developed to access the methylation status for a large number of genes or the entire genome. In this report, we describe the application of the Infinium® (East Sussex, UK) assay and BeadArray™ technology to the development of the Infinium Methylation assay, which enables a genome-wide high-throughput quantitative methylation profiling of the human genome.
Materials & methods
We analyzed 14 normal tissues, six human cancer cell lines and four B-lymphocyte noncancer cell lines. DNA from normal breast, ovary, prostate, kidney, liver, spleen, testis, stomach, lung, heart, brain, pancreas, skeletal muscle, colon and human cancer cell lines A431, HeLa, Jurkat, K-562, MCF-7 and Raji were purchased from BioChain Institute (CA, USA). DNA samples NA06999, NA07033, NA10923 and NA10924, were purchased from the Coriell Institute for Medical Research (NJ, USA).
Mung bean nuclease (Cat. No. M0250L) and CpG methyltransferase (M.SssI, Cat. No. M0226L) were purchased from New England BioLabs® (MA, USA). The EZ DNA Methylation™ kit for bisulfite conversion of genomic DNA (Cat. No. D5001) was from Zymo Research (CA, USA). REPLI-g® Mini Kit (Cat. No. 150025) from QIAGEN (Hilden, Germany) was used for the whole-genome amplification. We used the TOPO® TA Cloning® Kit for Sequencing (Cat. No. K457501, Invitrogen, CA, USA) for cloning and amplification of fragments used in bisulfite sequencing.
▪ Bisulfite conversion of genomic DNA
For bisulfite conversion, we used the EZ DNA Methylation kit from Zymo Research. We tracked the bisulfite conversion efficiency by monitoring BS-conversion controls on our Infinium Methylation 27k BeadChip. These controls were designed to monitor the conversion efficiency of a C to U base in a defined genomic HindIII site. This site was originally chosen to provide a gel-based quality control of bisulfite conversion efficiency. This site is flanked by PCR primer sites devoid of cytosines enabling an agnostic PCR amplification and simple HindIII restriction digest to evaluate BS conversion efficiency. Similarly, the Infinium query probe hybridized to a genomic sequence devoid of Cs. The genomic sequence is: AGATATGGGTATTATTTTGGAGAGCATAGGACTAGAATGTAATCaagcttGAGGAAGAGAGTAAAGAAATGGTGGAATGGAGATGATAG. The corresponding PCR primers are: (5´-AGATATGGGTATTATTTTGGAGA-3´ and 5´-CTATCATCTCCATTCCACCATT-3´), and the Infinium Methylation query probes are: TCTTTATACTATCATCTCCATTCCACCATTTCTTTACTCTCTTCCTCAA[A/G].
For optimized bisulfite conversion, we employed 500 ng of genomic DNA and followed the manufacturer’s protocol for the Zymo EZ DNA Methylation kit (kit #D5001) [102]. Namely, DNA was denatured by the addition of Zymo M-Dilution buffer (contains NaOH) and incubated for 15 min at 37°C. CT-conversion reagent (bisulfite-containing) was added to the denatured DNA and incubated for 16 h at 50°C in a thermocycler and denatured every 60 min by heating to 95°C for 30 s (please note: the manufacturer’s current protocol is based upon Illumina’s feedback to Zymo Research that bisulfite conversion efficiency can be improved by incorporating a cyclic denaturation protocol during the process of conversion).
After bisulfite conversion, the DNA was bound to a Zymo spin column and desulfonated on the column using M-desulfonation reagent per manufacturer’s protocol. The bisulfite-converted DNA was eluted from the column in 10 µl of elution buffer.
For the Infinium Methylation Assay, 4 µl of converted product (∼150 ng) was used in the whole-genome amplification (WGA) reaction. The Infinium Methylation Assay amplification and BeadArray protocols followed the Infinium Methylation Assay Experienced User Cards protocols. In brief, bisulfite-converted DNA was transferred to a new plate, denatured, neutralized and incubated at 37°C overnight for amplification. After amplification, the DNA was fragmented enzymatically, precipitated and resuspended in RA1 hybridization buffer. All subsequent steps were performed following the standard Infinium protocol. Fragmented DNA was dispensed onto the multisample HumanMethylation27 BeadChips, and hybridization performed in an Illumina Hybridization oven for 20 h. BeadChips were washed, primer extended, and stained per instructions. Finally, BeadChips were coated and then imaged on an Illumina BeadArray Reader. Images were processed with BeadStudio software (version 3.1.3.0) and methylation module (version 3.2.5) using the HumanMethylation27 270596 manifest (internal version).
▪ Generation of genomic DNA reference standards for methylation analysis
Unmethylated (U), hemi-methylated (H) and methylated (M) genomic reference standards were created from standard Coriell genomic DNA (gDNA; see Materials section). A total of 50 ng of gDNA (Coriell NA06999 and NA10924) was 100-fold amplified with the REPLI-g Mini Kit (Part No. 150023, QIAGEN) following manufacturer’s recommendations. Several reactions were carried out in parallel to generate enough material for the downstream processes. Amplified material was ethanol precipitated and re-suspended in TE buffer, followed by mung bean nuclease treatment to remove single-stranded DNA. Briefly, 2 µg of amplified DNA was incubated with 16 units of mung bean nuclease (New England Biolabs) in 1× NEB-2 buffer in a total reaction volume of 20 µl at 30°C for 1 h. The reaction was stopped by adding 0.5 M EDTA to 10 mM final concentration, heating to 65°C for 15 min and then ethanol precipitating. This whole-genome amplified, mung bean nuclease-treated NA06999 DNA served as the U reference. The NA10924 DNA was similarly processed and then methylated with M.SssI (CpG methyltransferase from New England Biolabs). Namely, 2 µg of whole-genome amplified, mung bean nuclease-treated NA10924 DNA was incubated with 6 units of M.SssI and 640 µM S-adenosylmethionine (SAM) in NEB-2 buffer (w/o MgCl2) in total reaction volume of 50 µl at 37°C for 2 h. 10× NEB-2 buffer consists of 500 mM NaCl, 100 mM Tris-Cl (pH 7.9), 10 mM MgCl2, and 10 mM dithiothreitol. M.SssI was inactivated by heating to 65°C for 15 min, and the sample was ethanol precipitated and re-suspended in TE buffer. The H reference was created by mixing U (NA06999) and M (NA10924) in a 1:1 stoichiometric ratio.
▪ Bisulfite sequencing
Methylation status of selected CpG sites was examined by bisulfite sequencing. Primers for the CD44 gene (forward: 5´-GAAAGGAGAGGTTAAAGGTTGAATT-3´; reverse: 5´- AATTTTAAAAAATAACAACCCTCCC-3´) were designed flanking the CpG sites of interest (cg08530414, cg17640322, cg01879488 and cg08606356) using MethPrimer software [103]. The PCR amplified fragments were cloned into the pCR4-TOPO Vector (Invitrogen) followed by transformation into Escherichia coli TOP10 competent cells (Invitrogen). Transformants containing recombinant plasmids were selected by blue/white colony screening. PCR inserts were directly amplified from the white colonies in the reaction mixture (35 µl) containing 3.5 µl GeneAmp 10× PCR buffer (Applied Biosystems), 1.5 units of AmpliTaq Gold® (Applied Biosystems), 1.5 mM MgCl2, 200 nM of dNTP and 200 nM each of M13 primers (Forward: 5´-GTAAAACGACGGCCAGT-3´ and Reverse: 5´-CAGGAAACAGCTATGAC-3´). The PCR reaction was subject to an initial heat denaturation step of 94°C for 10 min followed by 35 PCR cycles with each cycle consisting of 30 s at 94°C, 30 s at 50°C and 30 s at 72°C. After completion of the 35 amplification cycles, a final 5 min extension at 72°C was performed. The PCR products were sequenced by Agencourt Bioscience Corporation (MA, USA).
Results
▪ Array content selection
There are over 28 million CpG sites in the human genome. Using a set of empirical rules, we bioinformatically designed Infinium methylation probes to over 16 million of these CpG loci. For an initial demonstration of the Infinium Methylation assay, we selected a set of 27,578 CpG sites located within the proximal promoter regions (1 kb upstream and 500 bases downstream of transcription start sites (TSSs) of 14,475 consensus coding sequences (CCDS) genes and well-known cancer genes [104]. In addition, we included 254 assays across 110 miRNA promoters. On average, we selected two assays per CCDS gene and from 3–20 CpG sites for over 200 cancer-related and imprinted genes (Supplementary Table 1). Within promoter regions, assays were preferentially designed to sites within CpG islands whenever possible. We employed a NCBI ‘relaxed’ definition for CpG islands (CGIs) identified bioinformatically as DNA sequences (200 base window) with a GC base composition greater than 50% and a CpG observed/expected ratio [o/e] of more than 0.6 [23,24,105]. Using this relaxed definition, 60% of CCDS genes contain one or more CGI, and 40% contained no CGI.
▪ Probe design
There are several challenges in designing Infinium assay probes to query the state of a CpG site in bisulfite-converted DNA. Since most of the cytosines in the genome are converted to uracils, the uniqueness of any given sequence within the bisulfite converted genome decreases dramatically, potentially affecting specificity, and the fact that opposite strands are no longer complementary reduces the effective concentration of any given locus by a factor of 2. Nonetheless, we found that, in general, the specificity and sensitivity of the Infinium assay was sufficient to read out the requisite loci.
We adapted the Illumina Infinium I Whole Genome Genotyping (WGG) assay for measuring CpG methylation using quantitative ‘genotyping’ of bisulfite-converted genomic DNA. Bisulfite treatment of DNA converts unmethylated cytosines into uracil, and methylated cytosines remain unchanged. This C/T variant in the bisulfite-converted DNA can be queried using a standard methylation-specific assay design consisting of two probes per CpG locus: an ‘unmethylated’ and ‘methylated’ query probe (Figure 1). The 3´ terminus of the probe was designed to match either the protected cytosine (methylated design) or the uracil base resulting from bisulfite conversion (unmethylated design). The major challenge with locus-specific probe or array-based methylation assays is how to design the probe sequence to accommodate underlying CpG sites. For our current probe design, we assumed methylation is regionally correlated, and resolved underlying CpG sites to be in phase with the ‘methylated’ (cytosine) or unmethylated’ (uracil) query site [25]. The co-methylation assumption is based on the paper by Eckhardt et al. in which they bisulfite sequenced chromosomes 6, 20 and 22 [25,26]. Our probes have a span of 50 bases, and within this distance, methylation should be highly correlated. There are likely to be exceptions to this regional methylation rule. Nonetheless, although deviations from this hypothesis may affect the accuracy of the quantitative measurement, differential methylation measurements should still be valid.
▪ Methylation controls
To assess the overall functionality of the 27k assays and to generate a standard curve, we created three gDNA methylation reference standards: unmethylated (U), 50% hemi-methylated (H), and a 100% methylated (M) gDNA control. These three reference standards were created by in vitro demethylation (amplification-based) and methylation (M.SssI) of standard Coriell gDNA. Unmethylated gDNA was created by subjecting approximately 50 ng of Coriell gDNA to 100-fold whole-genome amplification (with Repli-G Mini kit) resulting in an output of 5–8 µg of amplified sample. Limiting the whole-genome amplification reaction to 100-fold amplification minimized representation bias (data not shown). The whole-genome amplified DNA from Coriell cell lines NA06999 and NA10924 was subjected to mung bean nuclease treatment to remove single-stranded DNA and create the unmethylated reference sample (U). The resultant NA10924 unmethylated DNA was treated with SssI methylase, which globally methylates all double-stranded CpG sites, to create a nearly completely methylated reference standard (M). The hemi-methylated reference standard was created by mixing equal proportions of U (NA0999) and M (NA10924) reference standards. These three validation standards were run on the Infinium 27k methylation array, and the corresponding methylation values (β = intensity[M]/(intensity[U] + intensity[M]) extracted. The distribution of β-values are consistent with the three reference standards with the unmethylated (U) standard showing low β-values, the hemi-methylated (H) standard showing intermediate β-values, and the methylated (M) standard having high β-values (Figure 2).
▪ Bisulfite conversion optimization
The Infinium assay employs whole-genome amplification and requires relatively intact DNA. Since all DNA present in the reaction will be amplified, it is important that bisulfite conversion is efficient yet minimizes DNA fragmentation. We evaluated several commercial bisulfite conversion kits and assessed the efficiency of conversion by monitoring a cytosine located in a HindIII site within the genome (see Methods section). We chose this particular HindIII site to enable a simple restriction enzyme (HindIII) digestion of a PCR product to monitor conversion efficiency. Furthermore, the PCR primers to this site were designed to be agnostic to the conversion efficiency. The upstream primer has no Gs, and the reverse complement downstream primer has no Cs. During the assay optimization we also monitored conversion efficiency by using a large set of probes complementary to bisulfite-converted DNA or unconverted gDNA.
We compared conversion efficiency between the standard protocol for the EZ DNA Methylation kit (Zymo Research) and the revised protocol using a precursor to the HumanMethylation27 array that contained a large number of probes to unconverted gDNA (data not shown). We also tested a number of other bisulfite conversion kits, but chose the optimized protocol for the EZ DNA Methylation kit based upon the array intensities and ratio of bisulfite-converted to unconverted signal intensities. The signal from the unconverted DNA may be elevated due to its enhanced amplification in the Infinium WGA assay, since more intact DNA amplifies more efficiently than degraded DNA.
In summary, we optimized the efficiency of the EZ DNA Methylation bisulfite conversion kit from Zymo Research using a cyclic denaturation protocol in which the gDNA was denatured at 95°C every hour for 30 s during the 16 h 50°C treatment process. Surprisingly, we found that this limited heat treatment did not significantly degrade the DNA relative to the non-heat denaturing treatment (data not shown). In general, bisulfite-treated DNA was relatively intact, ranging in size from a few hundred bases to several thousand bases, serving as a suitable substrate for the WGA reaction used in the Infinium assay. In the final product, we estimated that the conversion efficiency was over 99% by monitoring the ratio of the C to T signal from probes to the HindIII control site. The fact that this particular site converts with high efficiency does not suggest all Cs in the genome convert with similar efficiency. GC-rich regions that don’t denature as well may show poorer bisulfite conversion.
To test whether the presence of residual unconverted gDNA affects the assay, we spiked unconverted gDNA into bisulfite-converted DNA in various proportions (Figure 3). We computed the β-value and observed that for over 96.8% of the loci, the β-values were unaffected by the presence of unconverted DNA . However, we observed that for approximately 3.2% of the loci the β-values were sensitive to the presence of as little as 5% of unconverted DNA; thus, it is paramount to ensure the bisulfite conversion is complete to prevent these artifacts from occurring. Alternatively in future array designs, this spike experiment can be used to functionally screen for loci robust to trace amounts of unconverted DNA.
▪ Methylation status of 27k CpG sites across normal human tissues
To assess the biological performance of the assay, we analyzed the methylation profile of a panel of 14 different human tissues (male unless otherwise specified): brain (female), breast (female), colon, heart, kidney, liver, lung, ovary (female), pancreas (female), prostate, skeletal muscle, spleen, stomach and testis. In addition, we profiled four normal lymphoblastoid cell lines and six cancer cell lines of different origin (see Materials & methods section) (Supplementary Figure 1).
The average correlation R2 of β-values across a subset of 24 technical replicates was 0.992 (Figure 4A). Based upon the standard deviation of the β-values across the replicates and the average β-values of the control samples, we estimate a Δβ detection sensitivity of 0.2 (95% confidence level) across greater than 90% of the loci for any given pair of samples (Figure 4B). The Δβ sensitivity is higher at both the unmethylated state and highly methylated state. At unmethylated promoters, we can detect a change in β-value, on average, of approximately 0.07.
We evaluated the correlation of our Infinium Methylation assay with the absolute methylation state of a locus by comparing methylation β-values with methylation levels computed by clonal bisulfite sequencing across three genes and six samples. The Infinium Methylation results correlate favorably with bisulfite sequencing with an R2 of approximately 0.82 (Figure 5A). Similarly, we compared the correlation between GoldenGate® methylation and Infinium Methylation, showing an R2 of 0.86 (Figure 5B). In addition, we have compared results with data from over 2700 CpG sites on the same samples analyzed on the HumanMethylation27 BeadChip, and also sequenced on the Genome Analyzer (Illumina) using next-generation sequencing technology. We achieved correlation of β-values R2 of 0.85–0.87 (data not shown; manuscript in preparation). In another validation study, pyrosequencing was used for nine different genes across 72 tissue samples and three technical controls. The highly quantitative pyrosequencing data showed strong correlation with data obtained using the Infinium Methylation assay across the spectrum of β methylation values (average r2 = 0.91, range = 0.82–0.98) [106].
These results all indicate that the β-values from the Infinium Methylation assay reproducibly and accurately reflect the methylation state of the queried CpG loci.
We observed a significant difference in methylation states within a CGI versus outside of a CGI (Table 1 & Table 2, Supplementary Figure 2). In normal human tissues, the β average was 0.16 within a CGI and 0.58 outside of CGI (Figure 6A). The CpG sites within CGIs were largely unmethylated across all tissues (∼80% sites unmethylated, β < 0.2), whereas CpG sites outside of CGIs were moderately to highly methylated (only ∼12% sites unmethylated, β < 0.2) (Table 2). Within CGIs, only approximately 3–6% of the loci were highly methylated, in contrast to outside of CGIs where approximately 25–40% of loci were highly methylated. Moreover, tissue-specific methylation (variation in methylation across tissues) was much more prevalent in non-CGIs than within CGIs. The overall methylation level of CpG sites within a CGI, increased and outside of CGI slightly decreased, in several cancer cell lines that we analyzed for comparison, with the average β-value of 0.26 within a CGI and 0.53 outside of CGI (Figure 6B). In particular, the Raji cell line showed a gain of methylation across CpG sites and the K562 showed a loss of methylation. In general, only a small fraction of promoters/CGIs in tumor cells exhibit gain or loss of methylation. This is consistent with the change in distributions shown in Figure 6. In addition, Table 1 shows that methylation levels of CGI loci were higher on average in cancer cell lines versus normal tissues.
The definition of tissue-specific methylation is that certain loci are either methylated in some tissues and not in others or vice versa. To look for such loci, we filtered the data by looking for loci that exhibited a range greater than 0.4 and standard deviation greater than 0.15 across the 14 different tissues. These thresholds were set based upon maximal enrichment for X-chromosomal loci. We observed 504 CGI and 909 non-CGI sites out of 25,483 non-X chromosomal loci passing this threshold. This set was highly enriched for non-CGI loci, constituting 62.5% of the loci in the tissue-specific set versus 27.5% in the 27k set. Correspondingly, the average methylation level of this set was 0.47, in contrast to the global average of 0.15. This suggests that a significant fraction of the tissue-specific methylation occurs in non-CGI promoters. As a positive control, 19% of the loci on the X chromosome were contained in this enriched class, compared with 3.9% in the 27k set (females randomly inactivate one of the X chromosomes by methylating one of the parental chromosomes [27]). We also assayed the methylation status of 110 different miRNA promoters. Almost all miRNA promoters were unmethylated across the normal tissue panel. We observed that most miRNA promoters contained CGIs and were unmethylated. Only mir-254, mir-52, mir-135, mir-675 and mir-523 were highly methylated in normal tissues (Supplementary Figure 3).
▪ Methylation in tumor cell lines versus normal tissues
To illustrate the utility of differential methylation between tumor and normal samples, we examined the methylation state of four different classes of genes: germ-line specific genes [28]; Polycomb group (PcG) genes (targets of PRC2) [29]; Homeobox genes [30] and ribosomal housekeeping genes. We examined 55 germline-specific genes described by Koslowski et al.[28]. These are genes that are expressed only in germline cells and than repressed in somatic cells. Consistent with this classification, we observed that most genes were highly methylated across all normal tissues (Supplementary Figure 4). In contrast, the tumor cell lines exhibited a highly variegated expression pattern, many exhibiting loss of methylation. This is consistent with the global demethylation commonly observed in tumor cell lines. We examined a subset of 175 PcG target genes described by Widschwendter et al., and observed that most were relatively unmethylated in normal tissues but highly methylated in at least one or more tumor cell lines [29](Supplementary Figure 5). Similarly, the homeobox genes were mostly unmethylated in normal tissues, with only a small subset exhibiting moderate to full methylation (Supplementary Figure 6). In contrast a large fraction of unmethylated and intermediate methylated loci became fully methylated in one or more tumor cell lines. Interestingly, most homeobox genes fully methylated in normal tissues remained fully methylated in the tumor cell lines. The only exception was the K562 cell line, which exhibited demethylation in the PAX8, PAX4, CDX4, POUF6A, HOXA3, TGIF2LX and ALX3 genes. As a control, we examined the ribosomal housekeeping genes (RPL family) and observed minimal basal methylation across all normal tissues, as well as the cancer cell lines (Supplementary Figure 7). This observation is expected, since most housekeeping genes remain relatively unchanged during cancer cell growth and evolution.
Discussion
This is the first report of a scalable array-based genome-wide site-specific methylation assay that allows almost any set of CpG sites in the genome to be queried. We explored the use of the Infinium assay to read out the methylation state of CpG sites by generating a ‘pseudo-SNP’ via bisulfite conversion. Specifically, we demonstrate an array format supporting the analysis of over 27,000 loci across 12 samples on a single BeadChip. For this initial demonstration of the Infinium Methylation assay (design implemented in April 2007), we chose to analyze CpG sites in the proximal promoter of the CCDS gene set, particularly in CGIs near the TSS of CCDS genes, due to their potential biological significance [31].
In the future, the Infinium Methylation assay can be scaled to support over 4 million assays on a single BeadChip, and optimal array CpG content would ideally be generated by understanding which CpG sites are the most biologically informative, such as being highly correlated or anticorrelated with gene expression, histone marks, nucleosome positioning and so on. In addition, one can screen for differentially methylated CpG sites that serve as markers for a disease processes such as cancer. For instance if a CpG site is invariant across a large number of normal and tumor samples, it is probably not very useful site to include on the array. The goals of the HEP are to collect these methylation variable CpG sites, and upon completion of the project they can be included on a Beadchip, much like tag SNPs in our genotyping products.
One distinct advantage of the Infinium Methylation assay is that discrete CpG sites anywhere in the genome can be targeted in contrast to alternate array-based assays such as methylated-CpG island recovery assay (MIRA), methylated DNA immunoprecipitation (MeDIP), and methylation-sensitive restriction enzyme approaches that are limited to CpG-rich regions or restriction sites [15,16,32]. This is particularly limiting since many biologically important CpG sites may lie outside of CGIs. In a recent paper by Irizzary et al., they describe tissue specific methylation in CGI shores, regions flanking CGIs, but not within CGIs [33], whose methylation state is associated with evolutionary conservation, gene expression, sensitivity to demethylating agents and susceptibility to change in cancer. In a limited panel of three tissues including brain, liver and spleen, they identified over 16,000 tissue-specific differentially methylated regions. These CpG ‘poor’ regions could easily be adapted to the Infinium Methylation assay in future array designs.
Future perspective
The parallelization of both array and sequencing technology is having a transforming impact on both genome and epigenome science. In this report, we describe the use of highly parallelized Bead Array technology to measure CpG methylation states. However, next-generation sequencing technology is also enabling highly parallelized bisulfite-based genome-wide methylation analysis [34–37]. To date, most sequencing approaches have employed a restriction enzyme-based reduced representation approach that can query up to several million CpG sites; the caveat is that the reduced representation is biased to CpG-rich regions due to the use of CG-rich restriction sites. As an alternative, bisulfite shotgun sequencing of the entire genome would eliminate this bias, but currently the approach is relatively expensive and bioinformatically complex. Nonetheless, within the next several years, decreasing next-generation sequencing prices should enable the platform to serve as an extremely useful epigenomic discovery tool. By comparison, array-based methylation analysis offers several distinct advantages over sequencing, including: access to low-density CpG regions, reduced gDNA input requirements, simpler sample preparation protocols, lower running costs, higher sample throughput and simpler data storage and analysis. In summary, the Infinium Methylation assay is an ideal screening tool providing a powerful array-based assay for simple and rapid genome-wide methylation analysis of thousands to millions of CpG sites across large sample numbers. In conclusion, sequencing and array technologies can be effectively used in combination for various phases of an epigenome project, wherein sequencing might be applied to the discovery portion of a project and arrays applied to the screening portion.
| Tissue | CGI | Non-CGI |
|---|---|---|
| Brain | 0.144 | 0.579 |
| Breast | 0.190 | 0.571 |
| Colon | 0.168 | 0.602 |
| Heart | 0.137 | 0.593 |
| Kidney | 0.159 | 0.578 |
| Liver | 0.163 | 0.580 |
| Lung | 0.145 | 0.607 |
| Ovary | 0.167 | 0.560 |
| Pancreas | 0.163 | 0.554 |
| Prostate | 0.155 | 0.576 |
| Skeletal | 0.142 | 0.573 |
| Spleen | 0.155 | 0.619 |
| Stomach | 0.167 | 0.560 |
| Testis | 0.129 | 0.620 |
| Average normal | 0.156 | 0.584 |
| Hela | 0.200 | 0.509 |
| Raji | 0.389 | 0.645 |
| Jurkat | 0.305 | 0.597 |
| A431 | 0.257 | 0.507 |
| K562 | 0.173 | 0.282 |
| MCF7 | 0.259 | 0.652 |
Executive summary
▪ The Infinium® Methylation assay is a scalable, highly parallel, cost-effective approach to screening the state of tens of thousands to hundreds of thousands of CpG sites across dozens to thousands of samples.
▪ The current array assays over 27,000 CpG sites selected from proximal promoter regions of over 14,000 consensus coding sequences genes.
▪ Low-input gDNA requirements (∼500 ng), coupled with high reproducibility between technical replicates ( > 0.98 β R2) and high accuracy ( > 0.85 β R2) with bisulfite sequencing or GoldenGate® assay results are advantages of this technology.
▪ There are two probes per CpG assay. Probes query either ‘methylated’ or ‘unmethylated’ CpG state, with underlying CpG sites assumed to be in phase with each other.
▪ It is important to identify the biologically most informative CpG sites to generate optimal content for future versions of Infinium Methylation products.
Acknowledgements
We thank our Illumina colleagues in manufacturing for providing oligos and arrays for conducting the described research. We thank the BeadStudio team for the development of the Integrated Genome Viewer, and particularly Ivan Mikoulitch for development of the methylation module enabling analysis of the data. We also thank Eric Allen for helpful suggestions on data analysis.
Financial & competing interests disclosure
Marina Bibikova, Jennie Le, Bret Barnes, Shadi Saedinia-Melnyk, Richard Shen and Kevin L Gunderson are employees of Illumina and may own stock in the company. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was utilized in the production of this manuscript.
Bibliography
- 1 Bestor TH: Gene silencing. Methylation meets acetylation. Nature393,311–312 (1998).
- 2 Herman JG, Baylin SB: Gene silencing in cancer in association with promoter hypermethylation. N. Engl. J. Med.349,2042–2054 (2003).
- 3 Adorjan P, Distler J, Lipscher E et al.: Tumour class prediction and discovery by microarray-based DNA methylation analysis. Nucleic Acids Res.30,e21 (2002).
- 4 Bibikova M, Lin Z, Zhou L et al.: High-throughput DNA methylation profiling using universal bead arrays. Genome Res.16,383–393 (2006).
- 5 Clark SJ, Harrison J, Paul CL, Frommer M: High sensitivity mapping of methylated cytosines. Nucleic Acids Res.22,2990–2997 (1994).
- 6 Colella S, Shen L, Baggerly KA, Issa JP, Krahe R: Sensitive and quantitative universal Pyrosequencing methylation analysis of CpG sites. Biotechniques35,146–150 (2003).
- 7 Dupont JM, Tost J, Jammes H, Gut IG: De novo quantitative bisulfite sequencing using the pyrosequencing technology. Anal. Biochem.333,119–127 (2004).
- 8 Frommer M, McDonald LE, Millar DS et al.: A genomic sequencing protocol that yields a positive display of 5-methylcytosine residues in individual DNA strands. Proc. Natl Acad. Sci. USA89,1827–1831 (1992).
- 9 Gitan RS, Shi H, Chen CM, Yan PS, Huang TH: Methylation-specific oligonucleotide microarray: a new potential for high-throughput methylation analysis. Genome Res.12,158–164 (2002).
- 10 Singer-Sam J, LeBon JM, Tanguay RL, Riggs AD: A quantitative HpaII-PCR assay to measure methylation of DNA from a small number of cells. Nucleic Acids Res.18,687 (1990).
- 11 Kawai J, Hirotsune S, Hirose K, Fushiki S, Watanabe S, Hayashizaki Y: Methylation profiles of genomic DNA of mouse developmental brain detected by restriction landmark genomic scanning (RLGS) method. Nucleic Acids Res.21,5604–5608 (1993).
- 12 Khulan B, Thompson RF, Ye K et al.: Comparative isoschizomer profiling of cytosine methylation: the HELP assay. Genome Res.16,1046–1055 (2006).
- 13 Schumacher A, Kapranov P, Kaminsky Z et al.: Microarray-based DNA methylation profiling: technology and applications. Nucleic Acids Res.34,528–542 (2006).
- 14 Ordway JM, Budiman MA, Korshunova Y et al.: Identification of novel high-frequency DNA methylation changes in breast cancer. PLoS ONE2,e1314 (2007).
- 15 Weber M, Davies JJ, Wittig D et al.: Chromosome-wide and promoter-specific analyses identify sites of differential DNA methylation in normal and transformed human cells. Nat. Genet.37,853–862 (2005).
- 16 Rauch T, Pfeifer GP: Methylated-CpG island recovery assay: a new technique for the rapid detection of methylated-CpG islands in cancer. Lab. Invest.85,1172–1180 (2005).
- 17 Cheng YW, Shawber C, Notterman D, Paty P, Barany F: Multiplexed profiling of candidate genes for CpG island methylation status using a flexible PCR/LDR/Universal Array assay. Genome Res.16,282–289 (2006).
- 18 Bibikova M, Lin Z, Zhou L et al.: High-throughput DNA methylation profiling using universal bead arrays. Genome Res.16, (2006).
- 19 Gunderson KL, Steemers FJ, Lee G, Mendoza LG, Chee MS: A genome-wide scalable SNP genotyping assay using microarray technology. Nat. Genet.37,549–554 (2005).
- 20 Kuang SQ, Tong WG, Yang H et al.: Genome-wide identification of aberrantly methylated promoter associated CpG islands in acute lymphocytic leukemia. Leukemia22,1529–1538 (2008).
- 21 Omura N, Li CP, Li A et al.: Genome-wide profiling of methylated promoters in pancreatic adenocarcinoma. Cancer Biol. Ther.7,1146–1156 (2008).
- 22 Ordway JM, Bedell JA, Citek RW et al.: Comprehensive DNA methylation profiling in a human cancer genome identifies novel epigenetic targets. Carcinogenesis27,2409–2423 (2006).
- 23 Takai D, Jones PA: Comprehensive analysis of CpG islands in human chromosomes 21 and 22. Proc. Natl Acad. Sci. USA99,3740–3745 (2002).
- 24 Takai D, Jones PA: The CpG island searcher: a new WWW resource. In Silico Biol.3,235–240 (2003).
- 25 Eckhardt F, Lewin J, Cortese R et al.: DNA methylation profiling of human chromosomes 6, 20 and 22. Nat. Genet.38,1378–1385 (2006).
- 26 Rakyan VK, Hildmann T, Novik KL et al.: DNA methylation profiling of the human major histocompatibility complex: a pilot study for the human epigenome project. PLoS Biol.2,e405 (2004).
- 27 Li HP, Leu YW, Chang YS: Epigenetic changes in virus-associated human cancers. Cell Res.15,262–271 (2005).
- 28 Koslowski M, Sahin U, Huber C, Tureci O: The human X chromosome is enriched for germline genes expressed in premeiotic germ cells of both sexes. Hum. Mol. Genet.15,2392–2399 (2006).
- 29 Widschwendter M, Fiegl H, Egle D et al.: Epigenetic stem cell signature in cancer. Nat. Genet.39,157–158 (2007).
- 30 Holland PW, Booth HA, Bruford EA: Classification and nomenclature of all human homeobox genes. BMC Biol.5,47 (2007).
- 31 Illingworth R, Kerr A, Desousa D et al.: A novel CpG island set identifies tissue-specific methylation at developmental gene loci. PLoS Biol.6,e22 (2008).
- 32 Yan PS, Wei SH, Huang TH: Differential methylation hybridization using CpG island arrays. Methods Mol. Biol.200,87–100 (2002).
- 33 Irizarry RA, Ladd-Acosta C, Wen B et al.: The human colon cancer methylome shows similar hypo- and hypermethylation at conserved tissue-specific CpG island shores. Nat. Genet.41,178–186 (2009).
- 34 Meissner A, Gnirke A, Bell GW, Ramsahoye B, Lander ES, Jaenisch R: Reduced representation bisulfite sequencing for comparative high-resolution DNA methylation analysis. Nucleic Acids Res.33,5868–5877 (2005).
- 35 Meissner A, Mikkelsen TS, Gu H et al.: Genome-scale DNA methylation maps of pluripotent and differentiated cells. Nature454,766–770 (2008).
- 36 Smith ZD, Gu H, Bock C, Gnirke A, Meissner A: High-throughput bisulfite sequencing in mammalian genomes. Methods37(13),4331–4340 (2009).
- 37 Pomraning KR, Smith KM, Freitag M: Genome-wide high throughput analysis of DNA methylation in eukaryotes. Methods47,142–150 (2009).
- 101 Human Epigenome Project (HEP) website www.epigenome.org/
- 102 Instruction manual for the Zymo EZ DNA Methylation™ Kit www.zymoresearch.com/zrc/pdf/D5001i.pdf
- 103 MethPrimer design website www.urogene.org/methprimer/index1.html
- 104 CCDS Database website at NCBI www.ncbi.nlm.nih.gov/projects/CCDS/CcdsBrowse.cgi
- 105 NCBI MapViewer help document describing relaxed vs strict definition of CpG islands www.ncbi.nlm.nih.gov/projects/mapview/static/humansearch.html#cpg
- 106 Illumina’s iCommunity February 2008 newsletter. Describes application of the HumanMethylation27 BeadChip to the study of Barett’s esophagus DNA samples. The study also validates the performance of the Infinium Methylation assay using pyrosequencing www.illumina.com/icommunity/February2008/iC_INMethylationv7.pdf


(A) Distribution of unmethylated loci (β < 0.2) across normal tissues and tumor cell lines.
(B) Distribution of methylated loci (β > 0.75) across normal tissues and tumor cell lines.
CGI: CpG islands; Meth: Methylated; Unmeth: Unmethylated.





| Symbol | No. CpG sites | Chr. | Gene_ID | Synonym | Accession no. | Product |
|---|---|---|---|---|---|---|
| ABCB4 | 3 | 7 | GeneID:5244 | MDR3; PGY3; ABC21; MDR2/3; PFIC-3 | NM_018849.1 | ATP-binding cassette; subfamily B; member 4 isoform B |
| ALDH1A3 | 7 | 15 | GeneID:220 | ALDH6; RALDH3; ALDH1A6 | NM_000693.1 | Aldehyde dehydrogenase 1A3 |
| ALX4 | 23 | 11 | GeneID:60529 | FPP; PFM; PFM1; PFM2; KIAA1788; | NT_009237.17 | Aristaless-like homeobox 4 |
| ANP32E | 5 | 1 | GeneID:81611 | LANPL; LANP-L; MGC5350; | NM_030920.2 | Acidic (leucine-rich) nuclear phosphoprotein 32 family; member E |
| APBA1 | 4 | 9 | GeneID:320 | X11; X11A; MINT1; D9S411E; X11ALPHA | NM_001163.2 | Amyloid β A4 precursor protein-binding; family A; member 1 |
| APBA2 | 4 | 15 | GeneID:321 | X11L; MINT2; LIN-10; HsT16821; MGC99508; D15S1518E; MGC:14091 | NM_005503.2 | Amyloid β A4 precursor protein-binding; family A; member 2 |
| APC | 6 | 5 | GeneID:324 | GS; DP2; DP3; FAP; FPC; DP2.5 | NM_000038.3 | Adenomatosis polyposis coli |
| AR | 11 | X | GeneID:367 | KD; AIS; TFM; DHTR; SBMA; NR3C4; SMAX1; HUMARA | NM_000044.2 | Androgen receptor isoform 1 |
| ARF6 | 3 | 14 | GeneID:382 | NM_001663.2 | ADP-ribosylation factor 6 | |
| ASB4 | 4 | 7 | GeneID:51666 | ASB-4 | NM_145872.1 | Ankyrin repeat and SOCS box-containing protein 4 isoform b |
| ASPH | 3 | 8 | GeneID:444 | BAH; HAAH; JCTN; junctin; CASQ2BP1 | NM_020164.2 | Aspartate β-hydroxylase isoform e |
| ATG10 | 4 | 5 | GeneID:83734 | APG10L; pp12616; FLJ13954; DKFZP586I0418 | NM_031482.3 | APG10 autophagy 10-like |
| ATM | 5 | 11 | GeneID:472 | AT1; ATA; ATC; ATD; ATE; ATDC; TEL1; MGC74674; DKFZp781A0353 | NM_000051.3 | Ataxia telangiectasia mutated protein isoform 1 |
| ATP10A | 16 | 15 | GeneID:57194 | ATPVA; ATPVC; ATP10C; KIAA0566 | NM_024490.2 | ATPase; Class V; type 10A |
| ATRNL1 | 3 | 10 | GeneID:26033 | ALP; FLJ45344; KIAA0534; bA338L11.1; bA454H24.1; RP11-537G20.1 | NM_207303.1 | Attractin-like 1 |
| BCAP31 | 4 | X | GeneID:10134 | CDM; BAP31; 6C6-AG; DXS1357E | NM_005745.6 | B-cell receptor-associated protein 31 |
| BCDIN3 | 6 | 7 | GeneID:56257 | FLJ20257 | NM_019606.4 | Bin3; bicoid-interacting 3 |
| BCL2 | 15 | 18 | GeneID:596 | Bcl-2 | NM_000633.2 | B-cell lymphoma protein 2 α isoform |
| BIK | 6 | 22 | GeneID:638 | BP4; NBK; BBC1; BIP1 | NM_001197.3 | BCL2-interacting killer |
| BNC1 | 7 | 15 | GeneID:646 | BNC; BSN1; HsT19447 | NM_001717.2 | Basonuclin 1 |
| BRAF | 7 | 7 | GeneID:673 | BRAF1; RAFB1; B-raf 1; MGC126806; MGC138284 | NM_004333.2 | v-raf murine sarcoma viral oncogene homolog B1 |
| BRCA1 | 9 | 17 | GeneID:672 | IRIS; PSCP; BRCAI; BRCC1; RNF53 | NT_010755.15 | Breast cancer 1; early onset isoform BRCA1-δ15-17 |
| BTG4 | 4 | 11 | GeneID:54766 | PC3B; MGC33003 | NM_017589.2 | B-cell translocation gene 4 |
| C10orf4 | 5 | 10 | GeneID:118924 | FRA10AC1; F26C11.1-like | NM_203440.2 | FRA10AC1 protein isoform FRA10AC1-3.1 |
| C12orf24 | 4 | 12 | GeneID:29902 | HSU79274 | NM_013300.1 | Hypothetical protein LOC29902 |
| C12orf61 | 3 | 12 | GeneID:283416 | FLJ25590 | NM_175895.2 | Hypothetical protein LOC283416 |
| C19orf30 | 9 | 19 | GeneID:284424 | PGSF1; PGSF1a; PGSF1b | NM_174947.2 | Hypothetical protein LOC284424 |
| C1orf83 | 7 | 1 | GeneID:127428 | FLJ32112; FLJ39169; RP4-758J24.3 | NM_153035.1 | Hypothetical protein LOC127428 |
| CACNA1G | 6 | 17 | GeneID:8913 | NBR13; Ca(V)T.1; MGC117234 | NT_010783.14 | Voltage-dependent calcium channel α 1G subunit isoform 7 |
| CALCA | 8 | 11 | GeneID:796 | CT; KC; CGRP; CALC1; CGRP1; CGRP-I; MGC126648 | NT_009237.17 | Calcitonin isoform CALCA preproprotein |
| CALD1 | 3 | 7 | GeneID:800 | CDM; H-CAD; L-CAD; NAG22; MGC21352 | NM_033138.2 | Caldesmon 1 isoform 1 |
| CASP8 | 6 | 2 | GeneID:841 | CAP4; MACH; MCH5; FLICE; MGC78473 | NM_001228.3 | Caspase 8 isoform A |
| CAV1 | 6 | 7 | GeneID:857 | CAV; VIP21; MSTP085 | NT_007933.14 | Caveolin 1 |
| CCDC55 | 5 | 17 | GeneID:84081 | HSPC095; DKFZP434K1421 | NM_032141.2 | Hypothetical protein LOC84081 isoform 1 |
| CCNA1 | 3 | 13 | GeneID:8900 | NM_003914.2 | Cyclin A1 | |
| CCND1 | 18 | 11 | GeneID:595 | BCL1; PRAD1; U21B31; D11S287E; cyclin D1 | NM_053056.1 | Cyclin D1 |
| CCND2 | 13 | 12 | GeneID:894 | KIAK0002; MGC102758 | NM_001759.2 | Cyclin D2 |
| CD44 | 7 | 11 | GeneID:960 | IN; LHR; MC56; MDU2; MDU3; MIC4; Pgp1; CDW44; MUTCH-I; ECMR-III; MGC10468 | NM_000610.3 | CD44 antigen isoform 1 precursor |
| CDC7 | 6 | 1 | GeneID:8317 | Hsk1; CDC7L1; HsCDC7; huCDC7; MGC117361; MGC126237; MGC126238 | NM_003503.2 | CDC7 cell division cycle 7 |
| CDH1 | 8 | 16 | GeneID:999 | UVO; CDHE; ECAD; LCAM; Arc-1; CD324 | NM_004360.2 | Cadherin 1; type 1 preproprotein |
| CDH13 | 9 | 16 | GeneID:1012 | CDHH | NM_001257.3 | Cadherin 13 preproprotein |
| CDK2 | 7 | 12 | GeneID:1017 | p33(CDK2) | NT_029419.11 | Cyclin-dependent kinase 2 isoform 1 |
| CDKN1A | 6 | 6 | GeneID:1026 | P21; CIP1; SDI1; WAF1; CAP20; CDKN1; MDA-6; p21CIP1 | NM_078467.1 | Cyclin-dependent kinase inhibitor 1A |
| CDKN1C | 8 | 11 | GeneID:1028 | BWS; WBS; p57; BWCR; KIP2 | NT_009237.17 | Cyclin-dependent kinase inhibitor 1C |
| CDKN2A | 9 | 9 | GeneID:1029 | ARF; MLM; p14; p16; p19; CMM2; INK4; MTS1; TP16; CDK4I; CDKN2; INK4a; p14ARF; p16INK4; p16INK4a | NM_058195.2 | Cyclin-dependent kinase inhibitor 2A isoform 4 |
| CDKN2B | 10 | 9 | GeneID:1030 | P15; MTS2; TP15; INK4B | NM_004936.3 | Cyclin-dependent kinase inhibitor 2B isoform 1 |
| CHD2 | 3 | 15 | GeneID:1106 | DKFZp781D1727 | NM_001271.1 | Chromodomain helicase DNA binding protein 2 |
| CHEK2 | 3 | 22 | GeneID:11200 | CDS1; CHK2; LFS2; RAD53; HuCds1; PP1425 | NM_007194.3 | Protein kinase CHK2 isoform a |
| CHFR | 20 | 12 | GeneID:55743 | RNF116; RNF196; FLJ10796 | NM_018223.1 | Checkpoint with forkhead and ring finger domains |
| CLDND1 | 3 | 3 | GeneID:56650 | C3orf4; HSPC174; PRO6000; GENX-3745; DKFZP564P0462 | NM_019895.1 | Hypothetical protein LOC56650 |
| COPG2 | 4 | 7 | GeneID:26958 | 2-COP; FLJ11781 | NM_012133.2 | Coatomer protein complex; subunit γ 2 |
| CPA4 | 3 | 7 | GeneID:51200 | CPA3 | NM_016352.2 | Carboxypeptidase A4 preproprotein |
| CRABP1 | 7 | 15 | GeneID:1381 | RBP5; CRABP; CRABPI; CRABP-I | NM_004378.1 | Cellular retinoic acid binding protein 1 |
| CTDSP1 | 3 | 2 | GeneID:58190 | SCP1; NLIIF | NM_021198.1 | CTD (carboxy-terminal domain; RNA polymerase II; Polypeptide A) small phosphatase 1 isoform 1 |
| CTSZ | 7 | 20 | GeneID:1522 | CTSX | NM_001336.2 | Cathepsin Z preproprotein |
| CYP1A1 | 4 | 15 | GeneID:1543 | AHH; AHRR; CP11; CYP1; P1-450; P450-C; P450DX | NM_000499.2 | Cytochrome P450; family 1; subfamily A; polypeptide 1 |
| DAB2IP | 9 | 9 | GeneID:153090 | AIP1; AF9Q34; DIP1/2; KIAA1743 | NT_008470.18 | DAB2 interacting protein isoform 2 |
| DAPK1 | 8 | 9 | GeneID:1612 | DAPK; DKFZp781I035 | NM_004938.1 | Death-associated protein kinase 1 |
| DCC | 15 | 18 | GeneID:1630 | CRC18; CRCR1 | NT_010966.13 | Deleted in colorectal carcinoma |
| DDX17 | 4 | 22 | GeneID:10521 | P72; RH70; DKFZp761H2016 | NM_006386.3 | DEAD box polypeptide 17 isoform p82 |
| DIRAS3 | 14 | 1 | GeneID:9077 | ARHI; NOEY2 | NT_032977.8 | DIRAS family; GTP-binding RAS-like 3 |
| DLK1 | 4 | 14 | GeneID:8788 | FA1; ZOG; pG2; PREF1; Pref-1 | NT_026437.11 | δ-like 1 homolog isoform 2 |
| DLX5 | 18 | 7 | GeneID:1749 | NM_005221.4 | Distal-less homeo box 5 | |
| DNAJC18 | 6 | 5 | GeneID:202052 | MGC29463 | NM_152686.2 | DnaJ (Hsp40) homolog; subfamily C; member 18 |
| DNM2 | 7 | 19 | GeneID:1785 | DYN2; DYNII | NM_004945.2 | Dynamin 2 isoform 3 |
| DUS2L | 7 | 16 | GeneID:54920 | DUS2; SMM1; URLC8; FLJ20399 | NM_017803.3 | Dihydrouridine synthase 2-like (SMM1; S. cerevisiae) |
| DUSP1 | 7 | 5 | GeneID:1843 | HVH1; CL100; MKP-1; PTPN10 | NM_004417.2 | Dual specificity phosphatase 1 |
| DUSP4 | 4 | 8 | GeneID:1846 | TYP; HVH2; MKP2; MKP-2 | NM_057158.2 | Dual specificity phosphatase 4 isoform 2 |
| EDNRB | 8 | 13 | GeneID:1910 | ETB; ETRB; HSCR; ABCDS; HSCR2 | NT_024524.13 | Endothelin receptor type B isoform 2 |
| EFNB1 | 9 | X | GeneID:1947 | CFND; CFNS; EFL3; EPLG2; Elk-L; LERK2; MGC8782 | NM_004429.3 | Ephrin-B1 precursor |
| EML2 | 5 | 19 | GeneID:24139 | ELP70; EMAP2; EMAP-2 | NM_012155.1 | Echinoderm microtubule associated protein like 2 |
| EMR3 | 4 | 19 | GeneID:84658 | NM_032571.2 | Egf-like module-containing mucin-like receptor 3 isoform a | |
| ERBB2 | 11 | 17 | GeneID:2064 | NEU; NGL; HER2; TKR1; HER-2; c-erb B2; HER-2/neu | NM_004448.2 | erbB-2 isoform a |
| ESR1 | 7 | 6 | GeneID:2099 | ER; ESR; Era; ESRA; NR3A1; major ORF; DKFZp686N23123 | NM_000125.2 | Estrogen receptor 1 |
| EYA4 | 8 | 6 | GeneID:2070 | CMD1J; DFNA10 | NT_025741.14 | Eyes absent 4 isoform c |
| FANCF | 6 | 11 | GeneID:2188 | FAF | NM_022725.2 | Fanconi anemia; complementation group F |
| FBXO28 | 7 | 1 | GeneID:23219 | Fbx28; FLJ10766; KIAA0483 | NM_015176.1 | F-box protein 28 |
| FEN1 | 8 | 11 | GeneID:2237 | MF1; RAD2; FEN-1 | NM_004111.4 | Flap structure-specific endonuclease 1 |
| FHIT | 9 | 3 | GeneID:2272 | FRA3B; AP3Aase | NM_002012.1 | Fragile histidine triad gene |
| FLJ21106 | 3 | 4 | GeneID:80167 | NM_025097.1 | Hypothetical protein LOC80167 | |
| FLJ36046 | 3 | 22 | GeneID:164592 | NM_152612.2 | Hypothetical protein LOC164592 | |
| GALR1 | 7 | 18 | GeneID:2587 | GALNR; GALNR1 | NT_025004.13 | Galanin receptor 1 |
| GATA4 | 18 | 8 | GeneID:2626 | MGC126629 | NT_077531.3 | GATA binding protein 4 |
| GATA5 | 8 | 20 | GeneID:140628 | bB379O24.1 | NM_080473.3 | GATA binding protein 5 |
| GLMN | 4 | 1 | GeneID:11146 | GVM; GLML; FAB68; FAP48; FKBPAP; VMGLOM | NM_053274.1 | Glomulin isoform FAP68 |
| GLUL | 4 | 1 | GeneID:2752 | GS; GLNS | NM_001033056.1 | Glutamine synthetase |
| GNAS | 30 | 20 | GeneID:2778 | XL; AHO; GSA; GSP; POH; XL2; GPSA; NESP; GNAS1; PHP1A; PHP1B; GNASXL; NESP55; C20orf45; MGC33735; XLαs; dJ309F20.1.1; dJ806M20.3.3 | NT_011362.9 | Guanine nucleotide binding protein; α stimulating activity polypeptide 1 isoform a |
| GNMT | 7 | 6 | GeneID:27232 | NM_018960.4 | Glycine N-methyltransferase | |
| GPR153 | 4 | 1 | GeneID:387509 | PGR1; DKFZp762B2210 | NM_207370.1 | G protein-coupled receptor 153 |
| GPSN2 | 10 | 19 | GeneID:9524 | SC2; TER | NM_138501.4 | Glycoprotein; synaptic 2 |
| GPX3 | 5 | 5 | GeneID:2878 | NM_002084.2 | Plasma glutathione peroxidase 3 precursor | |
| GRB10 | 12 | 7 | GeneID:2887 | RSS; IRBP; MEG1; GRB-IR; KIAA0207 | NT_033968.5 | Growth factor receptor-bound protein 10 isoform a |
| GSTP1 | 7 | 11 | GeneID:2950 | PI; DFN7; GST3; FAEES3 | NT_033903.7 | Glutathione transferase |
| H19 | 16 | 11 | GeneID:283120 | ASM; BWS; ASM1; MGC4485; PRO2605; D11S813E | NR_002196.1 | – |
| HDAC11 | 7 | 3 | GeneID:79885 | FLJ22237 | NT_022517.17 | Histone deacetylase 11 |
| HIC1 | 3 | 17 | GeneID:3090 | hic-1; ZBTB29 | NM_006497.2 | Hypermethylated in cancer 1 |
| HOXA9 | 6 | 7 | GeneID:3205 | HOX1; ABD-B; HOX1G; HOX1.7; MGC1934 | NM_152739.2 | Homeobox protein A9 isoform a |
| HOXB4 | 8 | 17 | GeneID:3214 | HOX2; HOX2F; HOX-2.6 | NM_024015.3 | Homeo box B4 |
| HOXC5 | 3 | 12 | GeneID:3222 | CP11; HOX3; HOX3D | NM_018953.2 | Homeobox C5 |
| HOXD4 | 5 | 2 | GeneID:3233 | HOX4; HOX4B; HHO.C13; HOX-5.1; Hox-4.2 | NM_014621.2 | Homeobox D4 |
| HSD17B12 | 4 | 11 | GeneID:51144 | KAR | NM_016142.1 | Hteroid dehydrogenase homolog |
| HSPA2 | 8 | 14 | GeneID:3306 | NT_026437.11 | Heat shock 70kDa protein 2 | |
| HSPC268 | 8 | 7 | GeneID:154791 | NM_197964.1 | Hypothetical protein LOC154791 | |
| ICA1 | 3 | 7 | GeneID:3382 | ICA69; ICAp69 | NM_022307.1 | Islet cell autoantigen 1 isoform 1 |
| IGF2 | 5 | 11 | GeneID:3481 | FLJ44734 | NM_000612.2 | Insulin-like growth factor 2 |
| IGF2AS | 11 | 11 | GeneID:51214 | PEG8 | NT_009237.17 | Insulin-like growth factor 2 antisense |
| IGFBP3 | 5 | 7 | GeneID:3486 | IBP3; BP-53 | NT_007819.16 | Insulin-like growth factor binding protein 3 isoform a precursor |
| IKIP | 3 | 12 | GeneID:121457 | FLJ31051 | NM_201613.1 | IKK interacting protein isoform 3.1 |
| IMPDH1 | 5 | 7 | GeneID:3614 | IMPD; IMPD1; sWSS2608; DKFZp781N0678 | NM_183243.1 | Inosine monophosphate dehydrogenase 1 isoform b |
| INS | 4 | 11 | GeneID:3630 | NM_000207.1 | Proinsulin precursor | |
| ISYNA1 | 4 | 19 | GeneID:51477 | NM_016368.3 | Myo-inositol 1-phosphate synthase A1 | |
| ITPR2 | 9 | 12 | GeneID:3709 | IP3R2 | NT_009714.16 | Inositol 1;4;5-triphosphate receptor; type 2 |
| KCNQ1 | 23 | 11 | GeneID:3784 | LQT; RWS; WRS; LQT1; ATFB1; KCNA8; KCNA9; Kv1.9; Kv7.1; KVLQT1 | NT_009237.17 | Potassium voltage-gated channel; KQT-like subfamily; member 1 isoform 1 |
| KCNQ1DN | 6 | 11 | GeneID:55539 | BWRT; HSA404617 | NT_009237.17 | KCNQ1 downstream neighbor |
| KLK10 | 12 | 19 | GeneID:5655 | NES1; PRSSL1 | NM_002776.3 | Kallikrein 10 precursor |
| KRAS | 6 | 12 | GeneID:3845 | KRAS1; KRAS2; RASK2; KI-RAS; C-K-RAS; K-RAS2A; K-RAS2B; K-RAS4A; K-RAS4B | NM_004985.3 | c-K-ras2 protein isoform b |
| L3MBTL | 4 | 20 | GeneID:26013 | L3MBTL1; KIAA0681; H-L(3)MBT; dJ138B7.3; DKFZp586P1522 | NM_015478.4 | l(3)mbt-like isoform I |
| LOC129285 | 5 | 2 | GeneID:129285 | NM_152994.2 | Smooth muscle myosin heavy chain 11 isoform SM1-like | |
| LOC388152 | 4 | 15 | GeneID:388152 | MGC60197 | NM_203426.1 | Hypothetical protein LOC388152 |
| LOC51315 | 5 | 2 | GeneID:51315 | NM_016618.1 | Hypothetical protein LOC51315 | |
| LOX | 7 | 5 | GeneID:4015 | MGC105112 | NM_002317.3 | Lysyl oxidase preproprotein |
| MAGEL2 | 4 | 15 | GeneID:54551 | nM15; NDNL1 | NM_019066.2 | MAGE-like protein 2 |
| MEG3 | 7 | 14 | GeneID:55384 | NT_026437.11 | ||
| MEST | 10 | 7 | GeneID:4232 | PEG1; MGC8703; MGC111102; DKFZp686L18234 | NT_007933.14 | Mesoderm specific transcript isoform b |
| MGC33302 | 8 | 4 | GeneID:256471 | NM_152778.1 | Hypothetical protein LOC256471 | |
| MGMT | 26 | 10 | GeneID:4255 | NT_008818.15 | O-6-methylguanine-DNA methyltransferase | |
| MKRN3 | 4 | 15 | GeneID:7681 | D15S9; RNF63; ZFP127; ZNF127; MGC88288 | NM_005664.2 | Makorin; ring finger protein; 3 |
| MLH1 | 6 | 3 | GeneID:4292 | FCC2; COCA2; HNPCC; hMLH1; HNPCC2; MGC5172 | NT_022517.17 | MutL protein homolog 1 |
| MRPL12 | 8 | 17 | GeneID:6182 | 5c5-2; L12mt; MRPL7; RPML12; MGC8610; MRPL7/L12; MRP-L31/34 | NM_002949.2 | Mitochondrial ribosomal protein L12 |
| MSX1 | 18 | 4 | GeneID:4487 | HOX7; HYD1; OFC5 | NT_006051.17 | msh homeo box homolog 1 |
| MTHFR | 6 | 1 | GeneID:4524 | NT_021937.18 | 5,10-methylenetetrahydrofolate reductase (NADPH) | |
| MUC1 | 5 | 1 | GeneID:4582 | EMA; PEM; PUM; MAM6; PEMT; CD227; H23AG; mucin | NM_002456.4 | MUC1 mucin isoform 1 precursor |
| MYOD1 | 4 | 11 | GeneID:4654 | PUM; MYF3; MYOD | NM_002478.3 | Myogenic differentiation 1 |
| NDN | 6 | 15 | GeneID:4692 | HsT16328 | NM_002487.2 | Necdin |
| NEUROG1 | 6 | 5 | GeneID:4762 | AKA; ngn1; Math4C; NEUROD3 | NT_034772.5 | Neurogenin 1 |
| NNAT | 7 | 20 | GeneID:4826 | Peg5; MGC1439 | NM_005386.2 | Neuronatin isoform α |
| NR0B2 | 4 | 1 | GeneID:8431 | SHP; SHP1 | NM_021969.1 | Short heterodimer partner |
| OBFC2B | 7 | 12 | GeneID:79035 | MGC2731 | NM_024068.2 | Hypothetical protein LOC79035 |
| OSBPL5 | 8 | 11 | GeneID:114879 | ORP5; OBPH1 | NM_020896.2 | Oxysterol-binding protein-like protein 5 isoform a |
| OTUD4 | 4 | 4 | GeneID:54726 | HIN1; HSHIN1; KIAA1046; DKFZp434I0721 | NM_017493.4 | OTU domain containing 4 protein isoform 2 |
| OVOL1 | 4 | 11 | GeneID:5017 | HOVO1 | NM_004561.2 | OVO-like 1 binding protein |
| PCQAP | 3 | 22 | GeneID:51586 | TIG1; CAG7A; CTG7A; MED15; TIG-1; TNRC7; ARC105; DKFZp686A2214; DKFZp762B1216 | NM_015889.3 | Positive cofactor 2; glutamine/Q-rich-associated Protein isoform b |
| PDIK1L | 3 | 1 | GeneID:149420 | CLIK1L; RP11-96L14.4 | NM_152835.1 | PDLIM1 interacting kinase 1 like |
| PEG10 | 13 | 7 | GeneID:23089 | NT_007933.14 | Paternally expressed 10 | |
| PEG3 | 5 | 19 | GeneID:5178 | PW1; ZSCAN24; KIAA0287; DKFZp781A095 | NM_006210.1 | Paternally expressed 3 |
| PHLDA2 | 7 | 11 | GeneID:7262 | IPL; BRW1C; BWR1C; HLDA2; TSSC3 | NM_003311.3 | Pleckstrin homology-like domain family A member 2 |
| PIGO | 6 | 9 | GeneID:84720 | MGC3079; FLJ00135; MGC20536; DKFZp434M222; RP11-182N22.4 | NM_032634.2 | Phosphatidylinositol glycan; class O isoform 1 |
| PLAGL1 | 8 | 6 | GeneID:5325 | ZAC; LOT1; ZAC1; MGC126275; MGC126276; DKFZp781P1017 | NM_006718.2 | Pleiomorphic adenoma gene-like 1 isoform 2 |
| POLR2G | 6 | 11 | GeneID:5436 | RPB7; hRPB19; hsRPB7 | NM_002696.1 | DNA directed RNA polymerase II polypeptide G |
| POLR3D | 4 | 8 | GeneID:661 | RPC4; BN51T; TSBN51 | NM_001722.2 | RNA polymerase III 53 kDa subunit RPC4 |
| PPME1 | 9 | 11 | GeneID:51400 | PME-1 | NM_016147.1 | Protein phosphatase methylesterase-1 |
| PPP1R9A | 7 | 7 | GeneID:55607 | NT_007933.14 | Protein phosphatase 1; regulatory (inhibitor) subunit 9A isoform 1 | |
| PRDM2 | 4 | 1 | GeneID:7799 | RIZ; RIZ1; RIZ2; MTB-ZF; HUMHOXY1 | NM_012231.3 | Retinoblastoma protein-binding zinc finger protein isoform a |
| PRKCDBP | 4 | 11 | GeneID:112464 | SRBC; HSRBC; MGC20400 | NM_145040.2 | Protein kinase C; δ binding protein |
| PSMB6 | 6 | 17 | GeneID:5694 | Y; LMPY; DELTA; MGC5169 | NM_002798.1 | Proteasome β 6 subunit |
| PTEN | 8 | 10 | GeneID:5728 | BZS; MHAM; TEP1; MMAC1; PTEN1; MGC11227 | NT_030059.12 | Phosphatase and tensin homolog |
| PTGS2 | 6 | 1 | GeneID:5743 | COX2; COX-2; PHS-2; PGG/HS; PGHS-2; hCox-2 | NM_000963.1 | Prostaglandin-endoperoxide synthase 2 precursor |
| PTPNS1 | 3 | 20 | GeneID:140885 | BIT; MFR; P84; SIRP; MYD-1; SHPS1; SIRPA; CD172A; SHPS-1; SIRPα; SIRPα2; SIRP-ALPHA-1 | NM_080792.1 | Protein tyrosine phosphatase; nonreceptor type substrate 1 precursor |
| PTPRO | 9 | 12 | GeneID:5800 | PTPU2; GLEPP1; PTP-U2 | NM_002848.2 | Receptor-type protein tyrosine phosphatase O isoform b precursor |
| PWCR1 | 4 | 15 | GeneID:63968 | PET1; HBII-85 | NR_001290.1 | |
| PYCARD | 6 | 16 | GeneID:29108 | ASC; TMS1; CARD5; MGC10332 | NM_013258.3 | PYD and CARD domain containing isoform a |
| RAB32 | 6 | 6 | GeneID:10981 | NT_025741.14 | RAB32; member RAS oncogene family | |
| RARB | 4 | 3 | GeneID:5915 | HAP; RRB2; NR1B2 | NM_000965.2 | Retinoic acid receptor; β isoform 1 |
| RASSF1 | 9 | 3 | GeneID:11186 | 123F2; RDA32; NORE2A; RASSF1A; REH3P21 | NM_007182.4 | Ras association domain family 1 isoform A |
| RASSF5 | 9 | 1 | GeneID:83593 | RAPL; Maxp1; NORE1; RASSF3; MGC10823 | NT_021877.18 | Ras association (RalGDS/AF-6) domain family 5 isoform B |
| RB1 | 21 | 13 | GeneID:5925 | RB; OSRC | NT_024524.13 | Retinoblastoma 1 |
| RBP1 | 7 | 3 | GeneID:5947 | CRBP; RBPC; CRBP1; CRABP-I | NM_002899.2 | Retinol binding protein 1; cellular |
| RING1 | 4 | 6 | GeneID:6015 | RNF1 | NM_002931.3 | Ring finger protein 1 |
| RNF185 | 4 | 22 | GeneID:91445 | FLJ38628 | NM_152267.2 | Ring finger protein 185 |
| RNF41 | 4 | 12 | GeneID:10193 | NRDP1; SBBI03; MGC45228 | NM_005785.2 | Ring finger protein 41 isoform 1 |
| RUNX3 | 19 | 1 | GeneID:864 | AML2; CBFA3; PEBP2aC | NT_004610.18 | Runt-related transcription factor 3 isoform 2 |
| S100A9 | 3 | 1 | GeneID:6280 | MIF; NIF; P14; CAGB; CFAG; CGLB; L1AG; LIAG; MRP14; 60B8AG; MAC387 | NM_002965.2 | S100 calcium-binding protein A9 |
| SACM1L | 9 | 3 | GeneID:22908 | SAC1; KIAA0851; DKFZp686A0231 | NM_014016.2 | Suppressor of actin 1 |
| SEMA3B | 14 | 3 | GeneID:7869 | SemA; SEMA5; SEMAA; semaV; LUCA-1; FLJ34863 | NM_004636.2 | Semaphorin 3B isoform 1 precursor |
| SERPINB5 | 7 | 18 | GeneID:5268 | PI5; maspin | NT_025028.13 | Serine (or cysteine) proteinase inhibitor; clade B (ovalbumin); member 5 |
| SFRP1 | 5 | 8 | GeneID:6422 | FRP; FRP1; FrzA; FRP-1; SARP2 | NT_007995.14 | Secreted frizzled-related protein 1 |
| SFRP2 | 5 | 4 | GeneID:6423 | NT_016354.18 | Secreted frizzled-related protein 2 precursor | |
| SFRP4 | 6 | 7 | GeneID:6424 | FRP-4; FRPHE; MGC26498 | NT_007819.16 | Secreted frizzled-related protein 4 |
| SFRP5 | 5 | 10 | GeneID:6425 | SARP3 | NT_030059.12 | Secreted frizzled-related protein 5 |
| SFRS2 | 3 | 17 | GeneID:6427 | SC35; PR264; SC-35; SRp30b | NM_003016.2 | Splicing factor; arginine/serine-rich 2 |
| SGCE | 3 | 7 | GeneID:8910 | ESG; DYT11 | NM_003919.1 | Sarcoglycan; epsilon |
| SLC22A18 | 15 | 11 | GeneID:5002 | HET; ITM; BWR1A; IMPT1; TSSC5; ORCTL2; BWSCR1A; SLC22A1L; p45-BWR1A; DKFZp667A184 | NT_009237.17 | Tumor suppressing subtransferable candidate 5 |
| SLC5A8 | 7 | 12 | GeneID:160728 | AIT; MGC125354 | NM_145913.2 | Solute carrier family 5 (iodide transporter); member 8 |
| SMARCA3 | 4 | 3 | GeneID:6596 | HLTF; ZBU1; HLTF1; RNF80; HIP116; SNF2L3; HIP116A | NM_003071.2 | SWI/SNF-related matrix-associated actin-dependent regulator of chromatin a3 |
| SMPD3 | 7 | 16 | GeneID:55512 | NSMASE2; FLJ22593 | NM_018667.2 | Sphingomyelin phosphodiesterase 3; neutral membrane |
| SMPDL3A | 6 | 6 | GeneID:10924 | ASM3A; ASML3a; FLJ20177; yR36GH4.1 | NM_006714.2 | Acid sphingomyelinase-like phosphodiesterase 3A |
| SNRPN | 13 | 15 | GeneID:6638 | SMN; SM-D; RT-LI; HCERN3; SNRNP-N; SNURF-SNRPN | NT_026446.13 | Small nuclear ribonucleoprotein polypeptide N |
| SOCS1 | 8 | 16 | GeneID:8651 | JAB; CIS1; SSI1; TIP3; CISH1; SSI-1; SOCS-1 | NM_003745.1 | Suppressor of cytokine signaling 1 |
| SOCS2 | 14 | 12 | GeneID:8835 | CIS2; SSI2; Cish2; SSI-2; SOCS-2; STATI2 | NT_019546.15 | Suppressor of cytokine signaling-2 |
| SRF | 7 | 6 | GeneID:6722 | NM_003131.2 | Serum response factor (c-fos serum response element-binding transcription factor) | |
| STK11 | 4 | 19 | GeneID:6794 | PJS; LKB1 | NM_000455.4 | Serine/threonine protein kinase 11 |
| SUV420H2 | 3 | 19 | GeneID:84787 | MGC2705 | NM_032701.2 | Suppressor of variegation 4-20 homolog 2 |
| SYK | 7 | 9 | GeneID:6850 | NT_008470.18 | Spleen tyrosine kinase | |
| TCEB3C | 4 | 18 | GeneID:162699 | HsT829; TCEB3L2; MGC119353 | NM_145653.2 | Transcription elongation factor B polypeptide 3C |
| TFPI2 | 7 | 7 | GeneID:7980 | PP5; TFPI-2 | NM_006528.2 | Tissue factor pathway inhibitor 2 |
| TGFBR2 | 6 | 3 | GeneID:7048 | AAT3; MFS2; RIIC; HNPCC6; TGFR-2; TGFβ-RII | NT_022517.17 | TGF-β type II receptor isoform A precursor |
| THBS1 | 4 | 15 | GeneID:7057 | TSP; THBS; TSP1 | NM_003246.2 | Thrombospondin 1 precursor |
| THRB | 6 | 3 | GeneID:7068 | GRTH; THR1; ERBA2; NR1A2; THRB1; THRB2; ERBA-BETA; MGC126109; MGC126110 | NM_000461.2 | Thyroid hormone receptor; β |
| TIMP3 | 4 | 22 | GeneID:7078 | SFD; K222; K222TA2; HSMRK222 | NM_000362.4 | Tissue inhibitor of metalloproteinase 3 precursor |
| TMEM42 | 4 | 3 | GeneID:131616 | MGC29956 | NM_144638.1 | Transmembrane protein 42 |
| TNFRSF10C | 6 | 8 | GeneID:8794 | LIT; DCR1; TRID; CD263; TRAILR3 | NT_023666.17 | Tumor necrosis factor receptor superfamily; member 10c precursor |
| TNFRSF10D | 5 | 8 | GeneID:8793 | DCR2; CD264; TRUNDD; TRAILR4 | NM_003840.3 | Tumor necrosis factor receptor superfamily; member 10d precursor |
| TP53AP1 | 5 | 7 | GeneID:11257 | P53TG1; TP53TG1; P53TG1-D | NM_007233.1 | TP53 activated protein 1 |
| TP73 | 12 | 1 | GeneID:7161 | P73 | NM_005427.1 | Tumor protein p73 |
| TRA2A | 5 | 7 | GeneID:29896 | HSU53209 | NM_013293.2 | Transformer-2 α |
| TWIST1 | 7 | 7 | GeneID:7291 | SCS; ACS3; BPES2; BPES3; TWIST | NM_000474.3 | Twist |
| UBE3A | 8 | 15 | GeneID:7337 | AS; ANCR; E6-AP; HPVE6A; EPVE6AP | NM_130838.1 | Ubiquitin protein ligase E3A isoform 1 |
| UGDH | 8 | 4 | GeneID:7358 | GDH; UGD; UDPGDH; UDP-GlcDH | NM_003359.2 | UDP-glucose dehydrogenase |
| USP15 | 5 | 12 | GeneID:9958 | UNPH4; KIAA0529; MGC74854 | NM_006313.1 | Ubiquitin specific protease 15 |
| VHL | 7 | 3 | GeneID:7428 | RCA1; VHL1; HRCA1 | NM_000551.2 | Von Hippel-Lindau tumor suppressor isoform 1 |
| WDR37 | 7 | 10 | GeneID:22884 | FLJ40354; KIAA0982; RP11-529L18.2 | NM_014023.3 | WD repeat domain 37 |
| WDR8 | 3 | 1 | GeneID:49856 | FLJ20430; MGC99569 | NM_017818.2 | WD repeat domain 8 protein |
| WT1 | 20 | 11 | GeneID:7490 | GUD; WAGR; WT33; WIT-2 | NT_009237.17 | Wilms tumor 1 isoform C |
| ZIM2 | 12 | 19 | GeneID:23619 | NT_011109.15 | Zinc finger; imprinted 2 | |
| ZMYND10 | 4 | 3 | GeneID:51364 | BLU; FLU | NM_015896.2 | Zinc finger; MYND domain-containing 10 |
| ZNF148 | 3 | 3 | GeneID:7707 | BERF-1; BFCOL1; ZBP-89; ZFP148; pHZ-52; HT-BETA | NM_021964.1 | Zinc finger protein 148 (pHZ-52) |
| ZNF207 | 5 | 17 | GeneID:7756 | DKFZp761N202 | NM_001032293.1 | Zinc finger protein 207 isoform b |
| ZNF264 | 6 | 19 | GeneID:9422 | NM_003417.2 | Zinc finger protein 264 | |
| ZNF512 | 9 | 2 | GeneID:84450 | KIAA1805; MGC111046 | NM_032434.2 | Zinc finger protein 512 |
| ZNF688 | 12 | 16 | GeneID:146542 | NM_145271.3 | Zinc finger protein 688 isoform a |

