





























An integrated experimental and economic evaluation of cell therapy affinity purification technologies
Abstract
Aim: To present an integrated techno-economic analysis assessing the feasibility of affinity purification technologies using the manufacture of induced pluripotent stem cell-derived progenitor photoreceptors for retinal dystrophies as a case study. Materials & methods: Sort purity, progenitor yield and viable cell recovery were investigated for three cell sorting techniques: fluorescent-activated cell sorting (FACS); magnetic-activated cell sorting (MACS); and a novel technology SpheriTech beads. Experimentally derived metrics were incorporated into an advanced bioprocess economics tool to determine cost of goods per dose for each technology. Results & conclusion: Technical and bioprocess benefits were noted with SpheriTech beads which, unlike FACS and MACS, require no cell labeling. This simplifies the bioprocess, reduces cell loss and leaves target cells label free. The economic tool predicted cost drivers and a critical dose (7 × 107 cells per dose) shifting the most cost-effective technology from FACS to MACS. Process optimization is required for SpheriTech to compete economically.
First draft submitted: 1 November 2016; Accepted for publication: 11 April 2017; Published online: 16 June 2017
Induced pluripotent stem cell (iPSC)-derived products have a number of diverse applications ranging from drug screening to the study of disease mechanisms and clinical application as regenerative medicines [1–3]. However, the manufacture of target cell populations at a relevant scale and quality for clinical use remains a major challenge [4,5]. The production of this material requires efficient, scalable affinity purification processes for commercial manufacture of whole cell products [6]. As a nascent field, autologous therapies suffer from a lack of consensus to an optimal strategy for process scale-out. Decisional tools and economic approaches to bioprocess appraisal, which have been successfully applied in thefine-tipbiopharmaceutical sector [7–11], have the potential to aid process design and identify cost-effective scale-out strategies in this burgeoning industry. Studies have determined switch points (in terms of manufacture scale) whereby different cell culture platforms can be identified as the most cost-effective technology for expansion and differentiation of allogeneic and autologous therapies [12–14]. Further to this, large-scale platforms for iPSC-derived products have been assessed using economic parameters [15]. However, despite the breadth of cell therapies impacted, there have been no previous studies that examine and compare affinity purification strategies from an economic and operational standpoint on a quantitative basis. This paper presents an integrated techno-economic approach, whereby experimental data are fed into a bioprocess economics tool, to evaluate and compare the cost of goods (COG) associated with different high-resolution cell sorting techniques based on affinity purification.
For the treatment of retinal disorders, iPSCs must be reprogrammed from a donor biopsy, before a directed retinal differentiation is conducted to produce progenitor (also known as precursor) photoreceptors (Figure 1). Various studies have shown the potential of autologous cell therapies for the treatment of retinal diseases [3,16–18]. Retinal dystrophies describe a group of chronic degenerative conditions, characterized by degradation of photoreceptors, which result in visual impairment and frequently blindness. The most common, Retinitis pigmentosa, affects 1 in 4000 people worldwide and is linked to over 250 genes and 4500 different genetic defects with no current cure or proven treatment to slow disease progression [19]. Cell therapy as a treatment first showed promise when donor photoreceptors were transplanted into a retinal mouse model, then observed to maintain viability over 2 months [20]. However, the integration of transplanted cells to generate new functional synaptic connections was not observed until several years later, when progenitor photoreceptors were transplanted into adult mouse retinas. By injecting donor cells taken during peak rod genesis (postnatal days 3–5), progenitors were observed to fuse with the outer nuclear layer and restore vision [21,22]. As donor material is limited, retinal differentiation from a stem cell source has been employed to generate the required cellular material [23–25].

Flow sheet for autologous iPSC-derived progenitor photoreceptor cell therapy for the treatment of retinal dystrophy. The red box indicates the bioprocess unit operations costed in the computational model analysis.
iPSC: Induced pluripotent stem cell.
Affinity purification has become a core step in many therapies across a wide range of cell types and indications, owing to its ability to separate a target cell type from a heterogeneous population based on surface protein expression [26–31]. However, progenitor photoreceptors are predominantly characterized by the intracellular transcription factor CRX [32–34]. CRX has been used to isolate progenitor cells in a research setting by genetically modifying cell lines to co-express a fluorescent protein with CRX [35]. For antibody-dependent purification techniques in a clinical setting, genetically altering patient progenitor cells to fluoresce is impractical and adds significant cost and regulatory complications. As a result, a surface antigen is required for target cell identification. Marker CD73 has been well studied to characterize retinal photoreceptor progenitors in both mice and human retina [36–38]. Although CD73 is expressed in many different cell types, CD73-positive cell populations have successfully been used to select for CRX-positive cells in retinal cultures using affinity purification [36,39].
Fluorescent-activated cell sorting (FACS) and magnetic-activated cell sorting (MACS) are the current gold standard for affinity purification [40,41]. To investigate the effectiveness of CD73 as a marker, this paper examines these technologies to sort progenitor photoreceptors (Figure 2). For FACS, target cells are labeled with a fluorescent tag which is used to isolate them from heterogeneous suspensions. Instead of a fluorescent marker, MACS uses 50-nm paramagnetic beads as a method to bind and capture target cells. Once inside a strong magnetic field, target cells are held in place due to their bound paramagnetic beads.
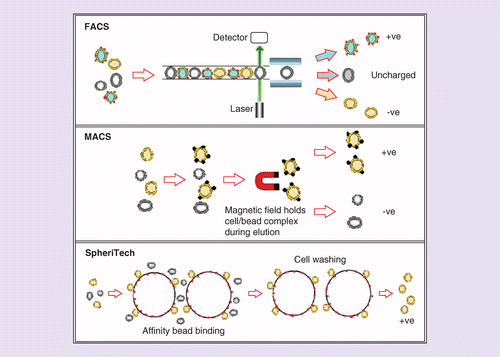
FACS uses a fluorescent tag to identify target cells in a single cell interrogation system before sorting. MACS requires paramagnetic beads to bind to target cells, which can then be held in place by a strong magnetic field for separation. SpheriTech purification works by cells binding to large polymer, antibody coated beads. They are eluted with trypsin and purified without retention of a cellular label.
FACS: Fluorescent-activated cell sorting; MACS: Magnetic-activated cell sorting.
In this paper, a novel affinity purification technology was also tested. SpheriTech beads are large hydrogel spheroids made with cross-linked poly-ϵ-lysine, to which antibody (CD73 in this case) has been covalently immobilized onto their surface. Similarly, to MACS beads, iron oxide has been infused during manufacture to make them paramagnetic. To purify target cells, beads are incubated with a heterogeneous cell suspension and, when target cells are bound, unwanted cells are eluted with beads held in place by a magnetic field. The beads are then washed, before CD73-positive captured cells are dissociated enzymatically by trypsinization. This means that, unlike FACS and MACS, there are no labeling steps which simplify the bioprocess and leave the target cell population free from any purification label. The performance of cell sorting with this composition of SpheriTech beads was tested against MACS and FACS, and their economic potential evaluated through the cost model.
To date, studies in this field have focused on either an economic [12–15] or experimental approach to bioprocess development in cell therapy. This paper presents, for the first time, an integrated experimental and economic appraisal of affinity purification platforms. The case study chosen represents one of the more challenging scenarios facing the cell therapy field, requiring the positive selection of iPSC-derived cells from potentially tumorigenic, diverse retinal and nonretinal cell impurities. Scenario analysis conducted in this study highlights which parameters have a critical impact upon bioprocess economics and operational feasibility. Although a single representative case study has been used, this methodology and the integrated techno-economic analysis can be applied to other bioprocesses and cell types that utilize affinity purification.
Materials & methods
Experimental materials & methods
Cell culture
Human pluripotent cells (MSUH001 iPSCs; Spanish Stem Cell Bank, Madrid, Spain) were cultured with inactivated mouse embryonic fibroblasts on 0.1% gelatin (Sigma-Aldrich, Dorset, UK) coated T25 flasks (Thermo Scientific, Leicestershire, UK) with media comprised of knockout DMEM, 20% (v/v) knockout serum replacement, 1% nonessential amino acids, 1 mM l-glutamine, 100 mM β-mercaptoethanol and 4 ng/cm3 bFGF (Invitrogen, Paisley, UK). iPSCs were incubated at 37°C and 5% (v/v) CO2 with media exchanged daily. Every 3–4 days, iPSCs were manually dissociated by surgical microdissection with afine-tip mini pastette (Alpha Laboratories, Hampshire, UK), then transferred to new mitomycin-inactivated mouse embryonic fibroblast flasks at a 1:3 ratio. Passages between p50 and p70 were used for experimentation and regularly tested for pluripotency via flow cytometry and immunostaining. Karyotype analysis was also carried out frequently to test for chromosomal abnormalities.
Human lung fibroblasts (MRC-5 cells [ATCC Catalog No. CCL-171]) were cultured in Nunc T175 flasks (Thermo Scientific) for a maximum of 40 population doublings in Eagle’s minimum essential medium (M5650; Sigma-Aldrich) with 10% fetal bovine serum (FBS; Seralab, West Sussex, UK), 1% glutamine and 1% MEM non-essential amino acids (Invitrogen).
Retinal differentiation
iPSCs were directed toward a retinal lineage via a modified Lamba differentiation protocol [23]. Undifferentiated iPSCs were enzymatically dissociated with 2 ml of Tryple (Invitrogen) for 5–10 min at 37°C. Cells were aggregated into embryoid bodies (EBs) in Aggrewell 400EX Plates (Stemcell Technologies, Grenoble, France) according to manufacturer’s guidelines, with 4.7 million cells seeded per well to form 4700 EBs with 1000 cells per EB. EB media comprised DMEM/F12 (Invitrogen), 10% (v/v) knockout serum replacement, 1 ng/ml of human recombinant DKK-1 (Cambridge Bioscience, Cambridge, UK), 1 ng/ml of human recombinant noggin (R&D Systems, MN, USA), 5 ng/ml of human recombinant IGF-1 (Miltenyi Biotec, Surrey, UK) and 1% (v/v) N-2 supplement (PAA Laboratories Ltd, Yeovil, UK).
On day 4, EBs were transferred into a 12-well plate coated in Matrigel (BD Bioscience, CA, USA) at a density of 30 EBs per well. Retinal differentiation media were changed every other day for 28 days. Media comprised DMEM/F12, 10 ng/ml of human recombinant DKK-1, 10 ng/ml of human recombinant Noggin, 10 ng/ml of human recombinant IGF-1, 1% (v/v) N-2 supplement, 2% (v/v) B27 supplement (PAA Laboratories) and 5 ng/ml bFGF.
On day 31, retinal differentiation cultures were dissociated using a combination of TrypLE and manual scraping with afine-tip mini pastette (Alpha Laboratories).
Cell counts
Cell counts were performed using a Neubauer-improved hemocytometer (Marienfeld-Superior, Lauda-Königshofen, Germany) with trypan blue (Sigma-Aldrich).
Flow cytometry & FACS
Flow cytometry was conducted according to the antibody manufacturer’s protocol (Miltenyi Biotec) with a few exceptions. Media were aspirated and cells enzymatically dissociated with 2 ml of Tryple for 10 min at 37°C and manual scraping with afine-tip mini pastette (Alpha Laboratories). The cell suspension was filtered through a 40-μm mesh, then quenched with blocking solution (5% [v/v] FBS in Dulbecco's phosphate-buffered saline; DPBS) and centrifuged for 5 min at 300 ×g to form a pellet. After aspirating the supernatant, the pellet was re-suspended in 100 mm3 of CD73 antibody (Miltenyi Biotec) per 107 nucleated cells, diluted to the manufacturer’s recommended concentration of 1:11. Samples were incubated for 20 min on ice in the dark, centrifuged and then re-suspended in 1 ml of blocking solution for flow cytometry or cell sorting.
Flow cytometry
Prepared samples were assessed using a flow cytometer (LSR II, 5 laser analyzer, BD Bioscience) and the data analyzed with FlowJo Version 10 (FlowJo LLC, OR, USA). A minimum of 50,000 events were initially gated based on forward and side scatter to remove cell debris and apoptotic cells, then doublets removed from forward scatter height against area. An isotype- or secondary-only control was run with the same gating strategy, and then positive expression was calculated from the top 1% of the control sample’s expression.
FACS
Prepared samples were sorted by a fluorescent-activated cell sorter (FACSAria II, BD Bioscience) for CD73 expression. For intracellular flow cytometry analysis after cell sorting, samples were fixed with 4% paraformaldehyde (PFA) on ice for 20 min, then permeabilized with 0.25% Triton X-100 solution for 10 min at 37°C. Cells were centrifuged to form a pellet, supernatant aspirated, and re-suspended in blocking solution for 30 min on ice. CRX (Insight Biotechnology Ltd, Wembley, UK) staining was then performed as described for the above extracellular protocol, with the antibody incubated for 20 min on ice at the manufacturer’s recommended concentration.
MACS
MACS was conducted according to the antibody manufacturer’s protocol (Miltenyi Biotec) with a few exceptions. Media were aspirated and cells enzymatically dissociated with 2 ml of TrypLE for 5–10 min at 37°C along with manual scraping using afine-tip mini pastette (Alpha Laboratories). The cell suspension was passed through a 40-μm sterile filter and quenched with buffer (5% [v/v] FBS in phosphate-buffered saline [PBS]), before being centrifuged for 5 min at 300 × g to form a pellet. The same CD73 antibody (as used for FACS and SpheriTech beads) was incubated with cells for 10 min in a fridge in the dark; 110 μl per 107 cells in a 1:10 ratio. An MS or LS column, dependent on scale (with a maximum total cell number of 2 × 108 or 2 × 109, respectively), was held in the MACS separator and rinsed with buffer. The cell suspension was applied and unlabeled cells collected below. The column was then removed and placed in another collection tube, before being flushed through with buffer using the supplied plunger and target cells collected.
SpheriTech bead manufacture
SpheriTech beads were manufactured and antibody-immobilized on the surface following protocols in the patent owned by SpheriTech [42]. About 10 g of Span80 (Sigma-Aldrich) was dissolved in 500 ml of toluene (Sigma-Aldrich) in a round bottomed flask. About 100 cm3 solution consisting of 10 g poly-ϵ-lysine, 3 g sebacic acid (Sigma-Aldrich) and 3 g N-methylmorpholine (Fisher Scientific, Leicestershire, UK) was prepared. About 1 g of iron (III) oxide magnetic NanoArc (Alfa Aesar, Lancashire, UK) was added, and the slurry was mixed into the round bottomed flask with 11.5 g of 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (Carbosynth, Compton, UK). After incubation for 2 h with agitation to allow for polymerization, the solvent layer was removed via filtration and particles were washed in a 500-μm sieve with excess water. Beads were stored in water with sodium azide (Sigma-Aldrich).
SpheriTech bead immobilization
Following details in the SpheriTech patent, 1 g of glutaric anhydride (Simga-Aldrich) was added to 1 cm3 of N-methylmorpholine (Fisher Scientific) in 2.5 cm3 of methanol (Sigma-Aldrich). SpheriTech beads were added to the vial and left on a roller for 24 h. Polymer beads were then washed with water, before incubation for 1 h in 170 mg of N-hydroxysuccinimide (VWR International, Leicestershire, UK) and 230 mg of 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride in pH 5 solution to activate carboxyl groups. The polymer was then washed with water and incubated with CD73 antibody (Miltenyi Biotec) overnight to give a final density of approximately 4 CD73 antibodies per μm2. Immobilized beads were washed with 5% v/v ethanolamine (Simga-Aldrich) to cap remaining free carboxyls. Beads were stored in sterile PBS before use.
SpheriTech cell sorting
Cells were suspended in PBS and incubated with CD73-immobilized beads at a density of around 1 × 106 per ml for 15 min on a roller. The beads were held in place with a magnet while unbound cells were eluted. Bound cells were dissociated using trypsin for 5–10 min at 37°C.
Confocal microscopy
Fluorescent confocal microscopy was used to visualize and count cells attached to beads. Cell suspensions were incubated with Red CMPTPX CellTracker dye (Thermo Scientific) for 30 min at room temperature. Cell sorting was then carried out as described above, and beads were imaged on an upright confocal microscope (TCS SPE confocal microscope; Leica, Milton Keynes, UK). Cells were counted on the visible half of the bead, and the value was multiplied by two to estimate total cells bound.
Scanning electron microscopy and immunogold staining
Scanning electron microscopy was conducted at the UCL Division of Biosciences Electron Microscopy facility using the Jeol 7401 high-resolution field emission scanning electron microscope.
For immunogold staining, SpheriTech beads were immobilized with excess CD73 antibody (Miltenyi Biotec) overnight. After washing with PBS, immobilized beads were incubated with secondary antibody conjugated with 20 nm colloidal gold (BioCell, West Lebanon, NH, USA) at a 1:20 ratio for 30 min at room temperature. A control, consisting of beads without CD73 immobilized antibody, was used and treated identically to the sample. Beads were washed with PBS and re-suspended in ethanol for critical point drying in a pressure vessel with liquid CO2. Samples were dried by evaporation then mounted and carbon-coated for electron microscopy.
For MRC-5 testing, cell suspensions were incubated with CD73 primary antibody for 20 min on ice at 1:50 ratio. Cells were fixed with 4% PFA for 10 min at room temperature before the same secondary antibody staining procedure was utilized as with SpheriTech beads, as well as for the control cell sample without primary CD73 staining. Samples were treated with 1% glutaraldehyde before treatment with 1% osmium for 20 min in the fridge. Samples were dehydrated in ethanol and then washed with hexamethyldisilazane (Sigma-Aldrich) before being mounted on a cover slip and being carbon coated.
Statistics
Three or more biological triplicates were tested for each experiment, and all values shown represent the mean with standard deviation as error bars. Statistical analysis was performed using GraphPad software with significance assessed through a two-tailed unpaired t-test where p ≤ 0.017.
Bioprocess economic tool methods
Tool definition
A bioprocess economics tool was developed to capture the COG associated with autologous human iPSC-derived cell therapy manufacture, when using either MACS, FACS or SpheriTech beads as a positive affinity purification. Experimentally derived data pertaining to the performance of different affinity purification platforms, as recorded using the methods outlined in the ‘Experimental materials and methods’ section, were fed into the tool as base-case assumptions. The tool consists of a cost model with mass balance, design, sizing, resource utilization and COG equations that was set up in Microsoft Excel (Microsoft, WA, USA), a database storing key data regarding different bioprocess technologies and iterative algorithms used during the sensitivity and scenario analyses that were developed in the Visual Basic for Applications plug-in (Microsoft).
COG model
The model was used to evaluate COG per dose for purification unit operations, as well as the bioprocess as a whole (detailed in Figure 1). Purification costs were calculated using the method described by Simaria et al. [12], originally used to calculate mesenchymal stem cell culture costs. Briefly, purification costs per dose are equal to the sum of annual material, labor and equipment depreciation costs associated with the purification operation, divided by the annual demand (number of doses per year) (Equation 1):
Material costs
Both FACS and MACS rely on fixed pieces of equipment; the SH800 cell sorter (Sony Biotechnology, Inc., Surrey, UK), FACSQuant Tyto (Miltenyi Biotec) or the CliniMACS system (Miltenyi Biotec). Fixed equipment is supported by disposable tubing and sorting components (MACS columns, FACS sorting chips), along with antibody-based buffers and reagents for each respective technology. Consumable list prices were obtained from BD Biosciences and Miltenyi Biotec for FACS, and Miltenyi Biotec for the MACS platform, to give a ‘price per run’ for the each sort (Cmat,r). A breakdown of these costs can be found in Supplementary Material 1 Material costs per dose (Cmat,d) for a given cell population input size, p-value, were calculated as follows (Equation 2), where uj is the number of units required to process a given cell population (according to the throughputs for a given technology found in Table 1):
| Purification method | Yield | Purity | Throughput (cells/h) | Turnaround (preparation and cleaning) | Differentiation efficiency | Dose size (cells) |
|---|---|---|---|---|---|---|
| MACS | 75.0 | 94.6 | 3.6 × 107 | 2.5 | 30% | 107 |
| FACS | 87.6 | 93.4 | 1.2 × 108 | 3.9 | – | – |

Labor costs
Labor costs associated with the cell sort were calculated from a basic operator’s annual salary. It was assumed that two operators would handle each unit at any time, as per Good Manufacturing Practice (GMP) requirements. Thus, the labor cost per dose associated with a given purification technology, Clab,d,j, was calculated as (Equation 3)
where wann is the annual salary for an operator, and demand represents the annual throughput of a facility in doses per year.
Equipment depreciation
Equipment depreciation costs were calculated by amortizing the total cost of any fixed equipment (Cequipment) required for purification over a 10-year period (y). Multiplication factors described in Jenkins et al. [13] were used to capture costs such as tax (ft), maintenance (fm) and insurance (fi) (Equation 4):
Indirect costs associated with purification were calculated according to the method in Simaria et al. [12], whereby indirect costs include equipment depreciation directly associated with the handling of disposable purification technologies.
Whole bioprocess COG
Purification costs were added to expansion and differentiation costs, calculated on the basis of a price per 107 cells and previous work carried out within UCL’s Advanced Centre for Biochemical Engineering (Jenkins et al. [13]; Weil BD, [Unpublished Data]). The whole bioprocess COG was calculated as follows (Equation 5):
where COGrep and COGexp are the respective COG per dose associated with iPSC reprogramming and iPSC cell culture, Pdiff represents the cell number required at the start of the differentiation to satisfy operation losses for a given dose size, and COGdiff is the COG per 107 cells associated with differentiation unit operations.
Sensitivity analysis
To determine key cost drivers associated with the bioprocess, process variables were altered by ±15% of their original value to assess the impact of best/worst case scenarios on COG per dose. For example, when the base-case differentiation efficiency of 30% was varied by ±15%, the worst case scenario value was 25.5%, and the best case value was 34.5%.
Scenario analyses
The cost model described above was used to provide a detailed cost breakdown, given base-case scale and performance characteristics, of different affinity purification technologies. In order to assess the impact of critical cost drivers, an algorithm was developed using the Visual Basic for Applications tool (Microsoft). By assigning incremental values to process parameters including dose size, differentiation efficiency, sort purity and purification yield, the most cost-effective, feasible technology across an array of scenarios was rapidly evaluated. Additionally, the algorithm was used to determine the purification yield at which SpheriTech beads became economically favored compared with FACS and MACS.
Results
Assessment of affinity purification technologies using iPSC-derived progenitor photoreceptors as a case study
A cell therapy for the treatment of retinal dystrophy was chosen to model the costs associated with purification and their impact upon the whole bioprocess. Starting with a human pluripotent stem cell source, stem cells were differentiated toward a retinal fate to produce progenitor photoreceptors. Progenitors were then purified using either MACS or FACS, and the (cell product and waste) output streams were analyzed by flow cytometry (Figure 3A). The surface marker CD73 was used to identify and sort progenitor photoreceptors during purification. A principle marker to define progenitors is the transcription factor CRX, and as such, the attributes of CD73 as a selection marker were assessed using CRX co-expression.

(A) Flow sheet detailing the experimental methods used to assess iPSC-derived, CD73 sorted cell populations for CRX expression by flow cytometry. (B) Comparison of CRX-positive expression (%) following CD73 purification with FACS or MACS in the positive and negatively sorted cell populations. n = 3 ± SD * = p ≤ 0.017. (C) Equations and calculation of progenitor yield and sort purity. Progenitor yield being the percentage of CRX-positive photoreceptors recovered through a CD73-positive sort; Sort purity being the percentage of cells within the CD73-positively sorted population which co-express CRX. (D) Viable cell recovery following FACS (n = 4) and MACS (n = 5) cell preparation and sorting. Viable cell recovery being the percentage of cells lost through respective cell (fluorophore or nano-bead) binding steps, and processing cells through a FACS machine or MACS column. (E) Protocol for FACS and MACS respective binding steps and cell processing, from which viable cell recovery was determined.
FACS: Fluorescent-activated cell sorting; iPSC: Induced pluripotent stem cell; MACS: Magnetic-activated cell sorting.
By analyzing the product and waste outputs, 93.4 and 94.6% of cells in the positive ‘cell product’ stream co-expressed CRX for MACS and FACS, respectively. This defines the sort purity, in other words, the number of CRX-positive progenitors within the CD73-positive sorted population. However, 12.5 and 4.2% of the corresponding negative ‘waste’ streams also co-expressed with CRX (Figure 3B). These losses are due to a difference between cell populations expressing CD73 and CRX, which require further characterization to define; this is beyond the scope of this paper. When taking into account the cell numbers per stream, a progenitor yield of 75.8 and 87.6% was calculated for MACS and FACS (Figure 3C).
The viable cell recovery from preparing cell suspensions and processing cells (as detailed in Figure 3D) was also investigated. The viable recovery was 58.7% for MACS and 69.6% for FACS, and these experimentally derived values were fed into the bioprocess economics tool.
Cost modeling of whole cell purification
The COG breakdown for individual components of the purification, as well as the impact of purification choice upon the entire bioprocess, was assessed using the bioprocess economics tool. Autologous therapies require a scale-out, rather than scale-up, strategy to be adopted for bioprocess design. It is unlikely that a single bioprocess facility could deal with an annual demand (doses per year) in excess of 10,000. Therefore, three different annual demands were initially trialed to evaluate the economies of scale that could be achieved.
At an annual demand of 1000 doses, purification costs were 12% greater with the use of MACS compared with FACS (Figure 4B) according to the tool. Disposable costs were 3.9-fold greater with the use of MACS. Although MACS offers a reduction in indirect, labor and reagent costs (39, 20 and 9%, respectively), these savings did not outweigh the stated difference in disposables.

(A) COG breakdown of the whole bioprocess by unit operation when FACS and MACS are employed as cell purification at annual demands of 100, 1,000, and 10,000 doses. (B) Breakdown of cell purification costs for the use of FACS and MACS when a dose size of 107 cells per patient, and a differentiation efficiency of 30% are assumed. The annual demand in this instance is 1,000 doses. A dose size of 107 cells per patient and a differentiation efficiency of 30% were assumed.
COG: Cost of goods; FACS: Fluorescent-activated cell sorting; iPSC: Induced pluripotent stem cells; QA: Quality assurance; QC: Quality control; MACS: Magnetic-activated cell sorting.
When looking at the entire bioprocess (Figure 4A), purification costs account for less than 11% of the total COG at high demands (1000+ doses per annum) regardless of the purification technology used, with differentiation accounting for approximately 50% of the total. However, performance characteristics of the purification step were observed to impact the required cell number to formulate the final dose, altering the costs associated with differentiation. To further investigate the impact that purification has upon bioprocess COG, sensitivity analysis was conducted by varying the tool’s base-case inputs by ±15% (Figure 5). Regardless of purification technology used, the key economic drivers (i.e., factors that have the greatest impact on COG per dose when varied) were all parameters which had a direct effect on the scale and cell number required for differentiation. Altering the progenitor yield and sort purity by 15% produced 21 and 16% swing in COG per dose for MACS (Figure 5A), and 20 and 17% swing for FACS, respectively (Figure 5B). These results appear to confirm that, while purification accounts for only approximately 10% of based-case total COG, its performance has a significant impact on the COG associated with the bioprocess.

Tornado charts showing the effects of varying key process parameters on the whole bioprocess COG per dose when (A) FACS or (B) MACS is used for cell purification. Best case and worst case values were calculated by adjusting each parameter by +/− 15% of the base case values given in Table 1, with an annual demand of 1,000 doses.
COG: Cost of goods; FACS: Fluorescent-activated cell sorting; MACS: Magnetic-activated sell sorting.
Scenario analyses
The impact of four key economic drivers on COG, as identified by the sensitivity analysis (namely, dose size, differentiation efficiency, sort purity and progenitor yield), were further explored in two-way scenario analyses by varying these factors from the base-case inputs in the bioprocess economics tool.
Final dose size was varied from 107 to 2 × 108 cells due to uncertainty in efficacious human dosage and scaling from preclinical work, while differentiation efficiency was assigned values between 30 and 80% to reflect the variation found in literature differentiation protocols [23,25,43–47]. At a differentiation efficiency of 30% (meaning after differentiation, 30% of the heterogeneous population are progenitor photoreceptors), FACS is the more cost-effective technology up to a dose size of 2.9 × 107 cells (Figure 6A). At differentiation efficiencies up to 80%, a maximum of 6.8 × 107 cells can be processed while keeping FACS cost effective. Exceeding these dose sizes produced an operating window whereby COGMACS < COGFACS.
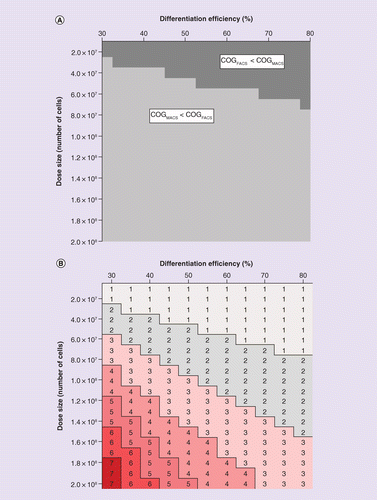
(A) Contour plot illustrating the effects of differentiation efficiency and dose size on whether FACS or MACS is the most cost-effective cell purification technology. Lightly-shaded areas indicate windows where FACS would prove more cost-effective than MACS. Darker-shaded areas indicate windows of operation whereby purification of target cells through MACS proves more cost-effective than FACS-based sorting. (B) Matrix illustrating the impact of dose size and differentiation efficiency on the number of FACS machines required to process an individual patient sample within a 4 h window. The numbers inside the matrix represent the number of FACS machines required to process the sample in each scenario.
COG: Cost of goods; FACS: Fluorescent-activated cell sorting; MACS: Magnetic-activated cell sorting.
At dose sizes above 7 × 107 cells, multiple FACS machines would be required to process an individual sample in 4 h. Operating above this restriction for a single-unit operation would significantly impact cell viability [48]. While the use of multiple FACS machines in parallel could theoretically be used to purify an individual cell suspension – with samples being pooled together after purification – it is an unlikely practice to be employed in a clinical setting. Thus, an operational window exists above dose sizes of 3 × 107 or 7 × 107 cells, depending on differentiation efficiency, where the use of FACS for purification is infeasible (Figure 6B).
Sort purity and progenitor yield were also assigned values between 60–100% and 85–100%, respectively, for MACS and FACS technologies (Figure 7A). When either factor fell below 80%, MACS-based purification costs (COGMACS PUR) were lower than FACS-based purification costs (COGFACS PUR). This correlates to the output from Figure 5, highlighting scale limitations that restrict FACS. To further illustrate this, sort purity was held at 95% while progenitor yield was change to either 75 or 85% (Figure 7B). At a progenitor yield of 85%, FACS purification costs are less than that of MACS purification due to reduced material costs (point B on Figure 7A). However, at a progenitor yield of 75% (point A on Figure 7A), FACS purification costs exceed MACS as higher cell numbers are required to satisfy the final dose size. The increase in labor costs associated with longer process times causes the COG per dose switch.

(A) Contour plot illustrating the effect of varying sort purity and progenitor yield on whether FACS or MACS is the most cost-effective purification technology. A dose size of 107 cells and annual demand of 1,000 doses are assumed. Points A and B represent the specific areas of the contour plot represented by the chart in Figure 7B. (B) Purification COG breakdown per dose for FACS and MACS purification when a yield of 75% or 85% is assumed with a fixed purity of 95% for both techniques (represented by Points A and B respectively in Figure 7A).
COGFACS PUR: Fluorescent-activated cell sorting-based purification costs; COGMACS PUR: Magnetic-activated cell sorting-based purification costs; FACS: Fluorescent-activated cell sorting; MACS: Magnetic-activated cell sorting.
Characterization of novel purification technology
To assess a novel purification technology against existing MACS and FACS techniques, SpheriTech beads were also used to isolate iPSC-derived progenitor cells. As part of the characterization of SpheriTech beads, scanning electron microscopy images were taken to visualize the topography of the polymer at different resolutions, comparable with how a cell or an antibody would interact with the surface (Figure 8A–C). The beads are smooth to the naked eye and on a cellular scale. At a submicrometer level, the beads are microporous, but this will have negligible effect on cell binding. Immobilized beads were cryosectioned to see if the antibody diffused into the core of a bead through these micropores during immobilization, but minimal diffusion was observed (data not shown).
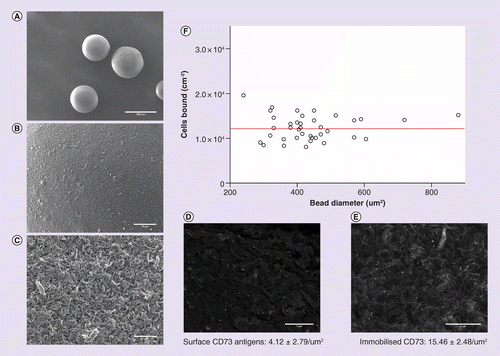
(A–C) Scanning Electron Microscopy images at three different resolutions to represent unmagnified visual inspection and the scale a cell or antibody would interact with the surface. Scale bars are 500 µm, 20 µm and 1 µm. (D) Immunogold staining of cell surface CD73 antigens (n = 12), scale bar 1 µm, and (E) CD73 antibody immobilised on SpheriTech beads (n = 12), scale bar 3 µm. (F) Scatter plot of cells bound/cm2 for a range of different SpheriTech bead sizes to determine the impact of bead diameter on binding by counting cells bound to SpheriTech beads via confocal microscopy. The red line is the mean cells bound/cm2: 1.24 × 104.
To assess surface antibody binding, immunogold staining was employed to label the covalently bound protein with gold nanoparticles for imaging. An excess antibody solution was used to fully saturate beads with maximum immobilized CD73. Electron microscopy images showed small clusters of the protein evenly distributed across the bead’s surface, with a density of 15.5 CD73/μm2 (Figure 8E). To determine the maximum required immobilized antibody density, cells were also stained and analyzed using the same methodology. CD73 antigens were labeled with the same immunogold antibody, and a density of 4.1 CD73/μm2 observed (Figure 8D). Consequently, an immobilized antibody density of approximately 4 μm-2 was used for further bead experimentation.
The impact of bead size upon cell binding density (cells/cm2) was investigated through both confocal microscopy cell counts and by calculating the difference between the starting cell number and supernatant (unbound) suspension after purification. MRC-5 were used as a convenient source of cells for this experiment to investigate the fundamentals of affinity bead binding. The cells express CD73 allowing the same antibody to be used in all studies. Between 200 and 1000 μm, bead size was observed to have no significant impact upon cell binding density (Figure 8F). Using visual counts from fluorescent confocal microscopy images of bound cells, an average density of 1.24 × 104 cells per cm2 was observed.
As a comparison to MACS and FACS, iPSC-derived progenitor photoreceptors were sorted using SpheriTech beads (Figure 9A). After dissociation, the heterogeneous retinal differentiation cell suspension was incubated with the affinity beads. Unbound cells were eluted, while bound cells were dissociated with trypsin and counted. The same methodology for cell counts, viable cell recovery, sort purity and progenitor yield calculations was used as previously applied to MACS and FACS. Additionally, the same CD73 antibody was used for all technologies. Performing flow cytometry analysis on the starting cells (postdifferentiation, dissociated cell suspension), supernatant and final dissociated cell populations following bead purification (Figure 9B) gave a sort purity of 89.8% and a yield of 54.9% (Figure 9D). The difference in cell number between the initial cell population and the sum of the supernatant and final dissociated cells together gave a viable cell recovery of 90.0% (Figure 9C).

(A) Protocol for SpheriTech binding steps and cell processing. (B) Bar chart showing the percentage of CRX expression in the starting cell population, the supernatant (unbound) cell suspension after elution, and the cell population dissociated from SpheriTech beads after binding. n = 6 ± SD. (C) Bar chart showing the viable cell count at each stage of processing cells during SpheriTech purification. Viable cell measurements were from haemocytometer counts with trypan blue (n = 6), except for the 'bound-to-beads' count which was derived from confocal microscopy images (n = 6). (D) Calculation of progenitor yield and sort purity using the same methodology as with FACS and MACS. Progenitor yield being the percentage of CRX-positive photoreceptors recovered after dissociating cells from SpheriTech beads after binding, compared to the starting cell population; Sort purity being the percentage of cells within the dissociated cell population which co-express CRX. Viable cell recovery was calculated from the supernatant and dissociated cell count combined as a percentage of the starting cell population.
FACS: Fluorescent-activated cell sorting; MACS: Magnetic-activated cell sorting.
Economic & operational feasibility of SpheriTech beads against gold standards
The breakdown of purification costs (Figure 4A) illustrates that reagents are the key cost driver for the unit operation. Specifically, the antibody cost was the greatest expenditure required for FACS and MACS, at 65 and 80% of the purification costs per dose, respectively (Table 2). This equates to around 6% of the COG for both operations. The antibody costs, as a percentage of the purification costs associated with SpheriTech, are a factor of 10 lower than current MACS and FACS. Here, antibody contributed only 4% of the purification costs. For the base-case bioprocess, the total COG associated with SpheriTech is £36,000 per dose, 5 and 7% higher than the COG per dose for MACS-based or FACS-based bioprocesses, respectively. The increase in COG is due to lower yields achieved through purification, which will be evaluated in the ‘Discussion’ section below. To directly assess performance characteristics, the purification yield for SpheriTech was assigned values between 50 and 100%, and compared against the COG per dose for current technologies at two different dose sizes: 107 and 108 cells (Figure 10). Intersects between the SpheriTech curves and base-case MACS and FACS costs indicate the switch points at which the COG per dose is equal (points B and E for MACS, point C for FACS). As a result, the quantitative improvement required by SpheriTech beads to compete with current purification methods is the distance between point B or C to point A for a dose size of 107 cells (increases of 5 and 8% points, respectively) or the distance between points D to E for a dose size of 108 cells (an increase of 6% points). As such, SpheriTech beads must operate with a yield greater than 60% for COGSpheriTech < COGMACS and 63% for COGSpheriTech < COGFACS at the base-case operating scale. With an increased dose size of 108 cells, an improvement in yield greater than 61% is necessary for COGSpheriTech < COGMACS.
| Purification method | Antibody cost per dose (GB£) | Percentage of purification cost (%) | Percentage of total cost (%) |
|---|---|---|---|
| FACS | 2098 | 80 | 6.2 |
| MACS | 2065 | 65 | 6.0 |
| SpheriTech beads | 49 | 4.2 | 0.15 |

Dose size of (A) 107 and (B) 108 cells. Point A and D represents the current SpheriTech bead yield. This is compared against the COG for MACS and FACS processes, given base case operating values (the dash and dotted horizontal lines). The table insert shows the percentage yield that must be achieved using SpheriTech beads to be economically favoured against MACS and FACS technology (which occurs at point B and C, respectively and at point E for MACS at the higher dose size).
COG: Cost of goods; FACS: Fluorescent-activated cell sorting; MACS: Magnetic-activated cell sorting.
Discussion
To critically assess affinity purification for cell therapy, a bioprocess economics tool was created using experimental data to analyze COG per dose for a bioprocess. The case study chosen was for an autologous retinal therapy with FACS and MACS purification technologies as this is an area of significant recent attention in the field of regenerative medicine.
A common problem with isolating rare subpopulations is the method to identify target cells. Antigen CD73 was used to positively select for progenitor photoreceptors, and feasibility of using this marker assessed through flow cytometry. A high sort purity for both MACS and FACS was noted; however, a significant difference was observed in progenitor yield. MACS relies on a binary selection method where magnetically labeled cells are held in place within a powerful magnetic field, while the remaining unlabeled cells are eluted as waste. As such, the degree of target protein expression is not a selection factor, and cell capture is dependent on the induced paramagnetic force from attached MACS beads holding the cell in place. As FACS interrogates each event independently to assess cell size (forward scatter) and granularity (side scatter), a full spectrum of protein expression can be observed which permits very lowly expressing cells to be positively identified and sorted. However, the cell preparation and physical cell loss from processing cells through a FACS machine is greater than that with MACS. Additionally, low throughput from single-cell interrogations increases processing time, and consequently limits operational feasibility at larger scales. As a result, there are compromises to be made for both purification methods; so economic feasibility is a key factor to distinguish them.
A dose size of 107 cells with 1000 annual doses was selected to perform cost breakdowns for each purification technique. At base-case inputs, both methods cost a little over GB£3000 per dose and were dominated by antibody-associated reagent costs. When looking at the full bioprocess, cell sorting corresponded to a relatively small percentage of the total COG, around 10–15%, with differentiation presenting the most significant contribution independent of annual doses. However, by combining an experimental and economic approach, it was noted that the demand on differentiation is directly influenced by performance of cell sorting which consequently impacts the process economics. The results of sensitivity analysis showed that, in addition to differentiation efficiency, the final dose size, sort purity and purification yield were also key economic drivers.
Currently, both dosage and differentiation efficiency are unknown as the therapy has only been tested in animal studies, so selecting a range to test was principally based on current literature and existing clinical therapies. Identification of the ‘true’ regenerative population to transplant into a patient is still a heavily researched topic, and consequently, operating at scales of 108 cells or more before purification is likely to be a necessity to select for a rare, subpopulation of cells. Several CRX antibodies have been shown to mark some bipolar cells, suggesting it is important to further characterize the target cell population using additional markers for photoreceptors, rods, cones Müller glia, horizontal and bipolar cells. Furthermore, iPSC therapies may not be truly autologous; through HLA matching, there is potential to treat multiple patients from one donor, dramatically increasing the production scale required.
These unknowns, in addition to effects regarding the integration efficiency of transplanted cells [22] and potentially increased cell numbers for quality control, dictated the chosen range examined of up to 108 cells per dose. FACS is a relatively low-throughput technology, and here it was found to only be suitable for therapies with a final dose size in the magnitude of 107 cells. A critical factor preventing the use of FACS at large scale is the need to split an individual patient sample between machines to limit process duration. MACS is more suited to larger scales by operating in a batch system which reduces capital expenditure and labor costs. Scenario analysis that assesses the impact of changing the sort purity and purification yield in Figures 6 & Figure 7 corroborates this finding: FACS is economically and practically favorable for the purification of small cell populations; however, when input cell populations increase, MACS is economically and operationally preferred.
The discussion has so far identified scale as the key element which dictates the choice of affinity purification; however, another critical feature with current technology is the requirement and retention of cellular labels. Both FACS and MACS require the cell product to be incubated with either fluorescent tags or iron oxide nanoparticles, before washing away unbound labels using centrifugation. This results in a more complex bioprocess, increasing cell loss and process risk by manipulating the cell product, while the cell label itself may also be unsuitable for transplantation [49–52]. As a result, SpheriTech beads were examined as they require no additional process steps and leave target cells label free. A label-free affinity purification method would be applicable across many regenerative therapies to simplify bioprocesses and reduce potential toxicological effects of cellular labels.
To determine the amount of antibody required for SpheriTech bead immobilization, immunogold staining was utilized to facilitate quantitative protein measurements on cell and bead surfaces. Once an optimum antibody immobilization density was found, the impact of bead diameter upon cell binding was addressed. Over 200 μm, the contact angle between cells and beads was found to plateau below 3° and, with increasing bead diameter, head toward an asymptote of 0° (Supplementary Material 2). As such, the contact angle is hypothesized to explain why the examined range of bead sizes did not impact cell binding per unit surface area. To ensure comparable binding characteristics between samples, a range of 200–400 μm diameter beads were used for further experimentation.
SpheriTech beads were then used to test differential binding of iPSC-derived progenitor photoreceptors, to provide a direct comparison with FACS and MACS. Flow cytometry analysis was again used to examine CRX and CD73 expressions within the positively and negatively sorted cell populations. Corresponding high purities (≥90%) were experienced for all technologies tested. As affinity cell sorting permits a high level of selectivity, and the same antibody was used with all three technologies, this was a hypothesized outcome. However, the progenitor yield was much lower with SpheriTech beads than with FACS and MACS.
For bigger beads, there is a compromise between maximizing the contact between cells and beads, while preventing clogging of the purification device. The reduced yield is likely due to target cells not coming into contact with beads during incubation or poor optimization of antibody binding, indicating this balance has not yet been optimized. The current proof-of-concept system shows that target cells can bind to SpheriTech beads and deliver label-free, high purity cellular products. However, future development must focus on engineering a device to improve cell contact with beads and optimize the chemistry of antibody immobilization to ultimately increase progenitor yield.
As the purification technologies considered in this paper vary from an industry gold standard to a proof-of-concept system susceptible to further development and optimization, we highlighted and quantified the improvement to SpheriTech beads in order for them to compete with current technologies. To quantify the improvement required for an economically favored process, the economics tool was used to determine the purification yield required for SpheriTech to match the COG per dose of FACS and MACS at base-case costs. Figure 10 highlights this critical yield threshold for SpheriTech to compete with current affinity technologies. Given a final dose of 107 cells, the technologies ranked FACS, MACS and then SpheriTech in order of economic preference, with a critical yield of 63% found for SpheriTech to compete (Figure 10A). If a higher dose of 108 cells is required, MACS becomes the preferred technology, followed by SpheriTech and then FACS (Figure 10B). At this dose, a critical yield of 61% was found for SpheriTech to beat the leading technology. These thresholds represent quantitative targets for future process development of SpheriTech beads. A similar approach could be applied to other novel technologies or a different cell bioprocesses to determine the optimization required.
The significant cost benefit with SpheriTech beads comes from the reduced antibody demand and higher viable cell recovery through bioprocess simplification. Consequently, although there is a difference in progenitor yield, a similar COG per dose is noted for all three purification platforms. As previously discussed, antibody cost is the key economic driver for both MACS and FACS operations at 65 and 80% of purification costs, respectively (Table 2). Unlike when MACS and FACS are used, concentration is not the critical factor for SpheriTech beads. Many MACS beads must bind per cell to elicit a sufficient paramagnetic charge for cell capture; numerous fluorophores must saturate suspended cells to produce an excited response and enable cell identification by FACS. For SpheriTech beads, only a low antibody concentration is needed to temporarily bind cells to the larger bead’s surface before elution. As a result, antibody demand is significantly lower accounting for just 4.2% of the purification costs, shifting focus to consumables as the key cost driver.
While purification costs only make up a small percentage of the COG per dose (Figure 4), sensitivity and scenario analysis illustrated that the performance characteristics of purification affect the cell number required for bioprocessing (Figures 5 & Figure 6). This significantly effects upstream processing COG. For our analysis, the same consumable demand was taken for MACS and SpheriTech bead operations, but a reduction in these costs would preferentially favor SpheriTech bead operations. Notably, viable cell recovery is much higher when using SpheriTech than with MACS and FACS due to the reduced number of processing steps during the purification operation; specifically, there are no cell labeling steps which reduce the number of cell washes needed and significantly improve cell recovery.
A further advantage to SpheriTech bead separations was observed during capture and elution steps. While MACS beads require a very strong magnetic field to induce a paramagnetic response, larger beads have a much lower iron oxide content but can be held in place with a weak magnetic field due to their size. This facilitates scaling up purification and, due to beads not constantly being held in suspension, permits other simple methods of physical bead capture that are not available to MACS. Furthermore, bead–cell complexes can be easily washed when target cells are bound. Although allogeneic therapies still present greater bioprocess and regulatory hurdles than autologous, large-scale purification techniques may play a key role in their future success. As SpheriTech bead purification is more favorable to commercially scaling operations than FACS or MACS, further scale-up work may be beneficial to the regenerative medicine field.
For both FACS and MACS, the purification label remains bound to the cellular product after sorting. For FACS, further development is still required to configure aseptic instruments for clinical use [53] and fluorescent material present in the retina could impact vision. For MACS, iron oxide has the potential to contribute to retinal degeneration [54], and nanobeads can be internalized by cells [55,56] causing cytotoxic effects which inhibit growth [57] or alter cell morphology [58], leading to severe complications [59,60]. A label-free, affinity purification method, which is economically competitive, could be a very clinically favorable alternative for many bioprocesses.
Conclusion
Affinity purification, based on the expression of surface markers, is the clinical gold standard for separating rare cell populations that cannot be crudely separated by density gradients. Despite their use in a wide variety of therapeutic applications, currently no study has been conducted into the quantitative impact that affinity purification technologies have upon a complete autologous bioprocess.
Here, we have illustrated an integrated techno-economic approach to the appraisal of different affinity purification technologies used in cell therapy bioprocesses. This study has highlighted and evaluated several key issues including the selection of surface markers for target cell isolation, the breakdown of purification costs and their impact upon the whole bioprocess through the creation of a bioprocess economics tool, and the key economic drivers and their impact upon the choice of purification technique.
This study found that FACS is limited in economic and operational feasibility by process scale. In this instance, at dose sizes below 7 × 107 cells, FACS was shown to be more cost effective than MACS. This boundary, however, is likely to be impacted by variations in yield and purity across different bioprocesses. At larger scales, an increase in the FACS COG per dose resulted in MACS becoming the most cost-effective technology.
While current purification methods require more complex bioprocesses and leave cells tagged after purification, SpheriTech beads offer a novel, scalable, label-free affinity purification solution with a simplified bioprocess. This technology could be of benefit to many therapies across the field of regenerative medicine by reducing the cost and bioprocess burden of cell purification. The beads were shown to differentially bind progenitor photoreceptors, eliciting high-purity cell sorts. If optimized to improve process yields, the bioprocess COG per dose would compete with FACS and MACS technologies across all scales tested.
Across the cell therapy industry, there is a growing need for scalable, label-free cell purification processes. Using an integrated technical and economic study has highlighted the impact that practical bioprocess considerations have upon the economic feasibility of a cell therapy. This proof-of-concept study with SpheriTech beads may have applications with adult stem cell, reprogrammed T cell and tissue engineered products.
Summary points
Operational characteristics of affinity purification technologies
The sort purity, progenitor yield and viable cell recovery of fluorescent-activated cell sorting (FACS) and magnetic-activated cell sorting (MACS) were experimentally determined using the surface marker CD73 to sort progenitor photoreceptors derived from human-induced pluripotent stem cells.
Bioprocess economics tool
Experimentally derived parameters were inputted into a bioprocess economic tool, and sensitivity analysis was conducted to determine the key cost drivers within purification and the bioprocess as a whole.
The impact of critical economic drivers was assessed across a range of scenarios, with zones of favorable operation established for MACS and FACS.
Characterization of novel purification technology
The novel affinity purification technology of SpheriTech beads was investigated and compared against FACS and MACS, as it offered label-free cells with a simplified bioprocess. The concentration of immobilized antibody and the impact of bead size upon cell binding were evaluated.
The operational characteristics of SpheriTech beads to purify induced pluripotent stem cell-derived progenitor photoreceptors were then experimentally established and compared with current affinity technologies.
Economic & operational feasibility of SpheriTech beads
The total bioprocess cost of goods associated with all three affinity technologies was determined with the economic tool. Direct comparisons were made between FACS, MACS and SpheriTech beads at dose sizes of 107 and 108 cells.
Conclusion
All affinity techniques tested produced high-purity separations; however, the progenitor yield and viable cell recovery varied between technologies.
Reagents, specifically antibody requirements, were the key cost associated with affinity purification.
FACS was found to be limited by throughput above a critical dose size of 7 × 107 cells per dose in this scenario, where MACS became the economically favored purification method.
While current affinity purification methods require additional cell-labeling process steps and leave cells tagged with fluorophores or paramagnetic beads after sorting, SpheriTech beads present a label-free solution to cell selection which simplifies the bioprocess.
For SpheriTech to compete economically with MACS and FACS, the progenitor yield must be optimized through additional process development.
Financial & competing interests disclosure
Financial support from the UK Engineering and Physical Sciences Research Council and SpheriTech Ltd. was gratefully acknowledged. University College London hosts the EPSRC Centre for Innovative manufacturing in Emergent Macromolecular Therapies with an academic network and a consortium of industrial and government users (Grant code: EP/G034656/1). The authors have no other relevant affiliations or financial involvement with an organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed. No writing assistance was utilized in the production of this manuscript.
Ethical conduct of research
The authors state that they have obtained appropriate institutional review board approval or have followed the principles outlined in the Declaration of Helsinki for all human or animal experimental investigations. In addition, for investigations involving human subjects, informed consent has been obtained from the participants involved.
Open access
This work is licensed under the Creative Commons Attribution 4.0 License. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
References
- 1 . Induced pluripotent stem cells for modelling human diseases. Philos. Trans. R. Soc. Lond. B., Biol. Sci. 366(1575), 2274–2285 (2011).
- 2 . Induced pluripotent stem cells for retinal degenerative diseases: a new perspective on the challenges. J. Genet. 88(4), 417–424 (2009).
- 3 . Current status of pluripotent stem cells: moving the first therapies to the clinic. Nat. Rev. Drug Discov. 14(10), 681–692 (2015).
- 4 . Large-scale expansion and exploitation of pluripotent stem cells for regenerative medicine purposes: beyond the T flask. Regen. Med. 7(1), 71–84 (2012).
- 5 . Human pluripotent stem cell-derived products: advances towards robust, scalable and cost-effective manufacturing strategies. Biotechnol. J. 10(1), 83–95 (2015).
- 6 . Bioprocessing challenges associated with the purification of cellular therapies. In: Stem Cells and Cell Therapy. Al-Rubeai M, Naciri M (Eds). Springer, Netherlands, 129–156 (2014).
- 7 . A tool for modeling strategic decisions in cell culture manufacturing. Biotechnol. Prog. 16(5), 829–836 (2000).
- 8 . Decision-support tool for assessing biomanufacturing strategies under uncertainty: stainless steel versus disposable equipment for clinical trial material preparation. Biotechnol. Prog. 21(2), 486–497 (2005).
- 9 . A prototype software methodology for the rapid evaluation of biomanufacturing process options. Biotechnol. Appl. Biochem. 48(2), 65–78 (2007).
- 10 . Decisional tool to assess current and future process robustness in an antibody purification facility. Biotechnol. Prog. 28(4), 1019–1028 (2012).
- 11 . Fed-batch and perfusion culture processes: economic, environmental, and operational feasibility under uncertainty. Biotechnol. Bioeng. 110(1), 206–219 (2013).
- 12 Allogeneic cell therapy bioprocess economics and optimization: single-use cell expansion technologies. Biotechnol. Bioeng. 111(1), 69–83 (2014).
- 13 . Patient-specific hiPSC bioprocessing for drug screening: bioprocess economics and optimisation. Biochem. Eng. J. 108, 84–97 (2015).
- 14 . Allogeneic cell therapy bioprocess economics and optimization: downstream processing decisions. Regen. Med. 10(5), 591–609 (2015).
- 15 . Design of large-scale manufacturing of induced pluripotent stem cell derived cardiomyocytes. Chem. Eng. Res. Des. 92(6), 1142–1152 (2014).
- 16 Stem cell based therapies for age-related macular degeneration: the promises and the challenges. Prog. Retin. Eye Res. 48, 1–39 (2015).
- 17 . Personalized therapeutic strategies for patients with retinitis pigmentosa. Expert Opin. Biol. Ther. 15(3), 391–402 (2015).
- 18 Transplantation of embryonic and induced pluripotent stem cell-derived 3D retinal sheets into retinal degenerative mice. Stem Cell Rep. 2(5), 662–674 (2014).
- 19 . A challenge to the striking genotypic heterogeneity of retinitis pigmentosa: a better understanding of the pathophysiology using the newest genetic strategies. Eye (Lond.) 30(12), 1542–1548 (2016).
- 20 . Reconstruction of degenerate rd mouse retina by transplantation of transgenic photoreceptors. Invest. Ophthalmol. Vis. Sci. 33(9), 2579–2586 (1992).
- 21 Retinal repair by transplantation of photoreceptor precursors. Nature 444(7116), 203–207 (2006).
- 22 Defining the integration capacity of ES cell-derived photoreceptor precursors. Stem Cells 30(7), 1424–1435 (2012).
- 23 . Efficient generation of retinal progenitor cells from human embryonic stem cells. Proc. Natl Acad. Sci. USA 103(34), 12769–12774 (2006).
- 24 Generation of Rx+/Pax6+ neural retinal precursors from embryonic stem cells. Proc. Natl Acad. Sci. USA 102(32), 11331–11336 (2005).
- 25 . Efficient stage specific differentiation of human pluripotent stem cells towards retinal photoreceptor cells. Stem Cells 30(4), 673–686 (2012).
- 26 . Chromatography of living cells using supermacroporous hydrogels, cryogels. Adv. Biochem. Eng. Biotechnol. 106, 101–127 (2007).
- 27 . High gradient magnetic cell separation with MACS. Cytometry 11(2), 231–238 (1990).
- 28 Clinical labeling and imaging of transplanted CD133+/CD34+ stem cells in patients with ischemic heart disease. Heart Surg. Forum 15(2), E116–E118 (2012).
- 29 . Continuous microfluidic immunomagnetic cell separation. Appl. Phys. Lett. 85(21), 5093 (2004).
- 30 Purification of CD4+ T cells for adoptive immunotherapy after allogeneic hematopoietic stem cell transplantation. Biol. Blood Marrow Transplant. 17(3), 374–383 (2011).
- 31 . Clinically relevant microfluidic magnetophoretic isolation of rare-cell populations for diagnostic and therapeutic monitoring applications. Anal. Chem. 84(3), 1336–1344 (2012).
- 32 . Expression of photoreceptor-associated molecules during human fetal eye development. Mol. Vis. 9, 401–409 (2003).
- 33 . Induction of functional photoreceptor phenotype by exogenous Crx expression in mouse retinal stem cells. Invest. Ophthalmol. Vis. Sci. 49(1), 429–437 (2008).
- 34 . Crx, a novel otx-like homeobox gene, shows photoreceptor-specific expression and regulates photoreceptor differentiation. Cell 91(4), 531–541 (1997).
- 35 . Derivation of traceable and transplantable photoreceptors from mouse embryonic stem cells. Stem Cell Rep. 2(6), 853–865 (2014).
- 36 CD73, a novel cell surface antigen that characterizes retinal photoreceptor precursor cells. Invest. Ophthalmol. Vis. Sci. 50(11), 5411–5418 (2009).
- 37 Transplantation of photoreceptor precursors isolated via a cell surface biomarker panel from embryonic stem cell-derived self-forming retina. Stem Cells 33(8), 2469–2482 (2015).
- 38 From confluent human iPS cells to self-forming neural retina and retinal pigmented epithelium. Proc. Natl Acad. Sci. USA 111(23), 8518–8523 (2014).
- 39 . Increased integration of transplanted CD73-positive photoreceptor precursors into adult mouse retina. Invest. Ophthalmol. Vis. Sci. 52(9), 6462–6471 (2011).
- 40 . Identification and isolation of hematopoietic stem cells. Arch. Med. Res. 34(6), 461–475 (2003).
- 41 . Embryonic stem cells : prospects for developmental biology and cell therapy. In Vitro 85(2), 635–678 (2005).
- 42 SpheriTech: WO2013041250 (2014).
- 43 Transplantation of adult mouse iPS cell-derived photoreceptor precursors restores retinal structure and function in degenerative mice. PLoS ONE 6(4), e18992 (2011).
- 44 Toward the generation of rod and cone photoreceptors from mouse, monkey and human embryonic stem cells. Nat. Biotechnol. 26(2), 215–224 (2008).
- 45 . Induced pluripotent stem cell technology for generating photoreceptors. Regen. Med. 6(4), 469–479 (2011).
- 46 . The role of endothelial cells in the retinal stem and progenitor cell niche within a 3D engineered hydrogel matrix. Biomaterials 33(21), 5198–5205 (2012).
- 47 Optic vesicle-like structures derived from human pluripotent stem cells facilitate a customized approach to retinal disease treatment. Stem Cells 29(8), 1206–1218 (2011).
- 48 . The impact of manual processing on the expansion and directed differentiation of embryonic stem cells. Biotechnol. Bioeng. 99(5), 1216–1229 (2008).
- 49 . Potential toxicity of superparamagnetic iron oxide nanoparticles (SPION). Nano Rev. 1(0), 1–15 (2010).
- 50 Magnetite- and maghemite-induced different toxicity in murine alveolar macrophage cells. Arch. Toxicol. 88(8), 1607–1618 (2014).
- 51 . Potential toxic effects of iron oxide nanoparticles in in vivo and in vitro experiments. J. Appl. Toxicol. 32(6), 446–453 (2012).
- 52 Toxicity of organic fluorophores used in molecular imaging: literature review. Mol. Imaging 8(6), 341–354 (2009).
- 53 Sterile and disposable fluidic subsystem suitable for clinical high speed fluorescence-activated cell sorting. Cytometry B. Clin. Cytom. 70(5), 344–354 (2006).
- 54 Iron homeostasis and toxicity in retinal degeneration. Prog. Retin. Eye Res. 26(6), 649–673 (2007).
- 55 . Mechanisms of nanoparticle internalization and transport across an intestinal epithelial cell model: effect of size and surface charge. Mol. Pharm. 11(12), 4363–4373 (2014).
- 56 Quantitative analysis of nanoparticle internalization in mammalian cells by high resolution x-ray microscopy. J. Nanobiotechnology 9, 14 (2011).
- 57 . Nanotechnology and health safety – toxicity and risk assessments of nanostructured materials on human health. J. Nanosci. Nanotechnol. 7(9), 3048–3070 (2007).
- 58 . New findings about iron oxide nanoparticles and their different effects on murine primary brain cells. Int. J. Nanomedicine 10, 2033–2049 (2015).
- 59 In vitro effects of nanoparticles on renal cells. Part. Fibre Toxicol. 5, 22 (2008).
- 60 . Cellular uptake of nanoparticles as determined by particle properties, experimental conditions, and cell type. Environ. Toxicol. Chem. 33(3), 481–492 (2014).

