





























Programmed cell death-1/programmed cell death ligand-1 checkpoint inhibitors: differences in mechanism of action
Abstract
Programmed cell death-1 (PD-1)/programmed death-ligand 1 (PD-L1) checkpoint inhibitors are widely used in many types of solid tumors, and are often considered to be in the same immunotherapy subclass. This review explores whether specific agents in these two categories exhibit differences in their mechanism of action, pharmacokinetics and pharmacodynamics, and clinical efficacy and safety. Due to the complicated functional pathways in the immune checkpoint system, the epitopes, interfaces and signal pathways between PD-1: PD-L1/PD-L2, PD-L1/CD28/CTLA-4: B7-1 axes often overlap and affect each other. Therefore, the mechanisms of action of PD-1 and PD-L1 inhibitors reflect the corresponding cross connectivity and their unique characteristics. Only head-to-head comparative studies can provide definitive information regarding clinical efficacy and safety differences between specific PD-1/PD-L1 inhibitors.
Programmed cell death-1 (PD-1)/programmed death-ligand 1 (PD-L1) inhibitors are commonly considered as being in the same subclass of immunotherapeutic agents and are widely used in malignant melanoma (MM), non-small-cell lung cancer (NSCLC) and many other types of tumors. However, there is considerable interest among researchers as to whether specific agents in these two categories exhibit differences in their mechanism of action, pharmacokinetic and pharmacodynamic characteristics, clinical efficacy, and safety. This review analyzes the differences between agents in each category and the related underlying mechanisms, and explores whether these differences can contribute to differences in clinical efficacy and safety, and could thus provide some basis for clinical decision making and patient selection.
Mechanism of action
Targeted signaling
PD-1 inhibitor: PD-1/PD-L1 & PD-1/PD-L2 axis
PD-1 is a key immune checkpoint receptor, which mainly expresses on activated T, B, dendritic (DCs), natural killer (NK) and Treg cells. On the surface of activated T cells, PD-1 expression is upregulated after the recognition of peripheral antigens by T cells; subsequently, the elevated binding of PD-1 to PD-L1 and PD-L2 becomes a key step for downstream inhibitory signaling [1]. PD-1 is also associated with increased Treg-cell proliferation and enhanced immunosuppressive function (Figure 1).

Reproduced with permission from [2] © Frontiers in Pharmacology (CC BY 4.0) (2017).
A series of inflammatory cytokines, such as IFN-γ, which is secreted by activated T and NK cells, can promote PD-L1 expression in a variety of cell types, including epithelial, endothelial, hematopoietic and tumor cells. Oncogenes may also play an important role in stimulating the expression of PD-L1. In contrast, PD-L2 is expressed mainly in activated DCs, macrophages, bone marrow-derived mast cells, peritoneal B1 cells, helper T (Th) 2 cells and intrapulmonary nonhematological cells. Similar to PD-L1, PD-L2 expression can be observed in some types of tumor cells. PD-L1 and PD-L2 expression is correlated in tumors; however, in some types of tumor cells, PD-L1 can be present in the absence of PD-L2, and vice versa [3,4]. After the recognition of tumor antigens by peripheral T-cell receptors (TCRs), PD-L1 or PD-L2 will bind to PD-1 and activate downstream-related signaling pathways, block TCR signaling via feedback inhibition, and downregulate the expression of specific antiapoptotic molecules including Bcl-xL and proinflammatory factors, ultimately inhibiting T-cell survival, proliferation and immune function. In addition, the interaction of PD-1 with its ligand can also affect the cell cycle (by increasing the expression of p15 decreasing the transcription level of SKP2, preventing T cells from entering the G1 phase, and eventually inhibiting cell proliferation), stimulate tumor-specific T-cell apoptosis and promote the differentiation of Th cells into Tregs [5].
Although the role of PD-L1 has been evaluated more thoroughly by clinical researchers, and PD-L1 has also been used more widely in clinical settings, PD-L2 also plays an important role in the negative regulation of T cells, one of the necessary conditions that lead to immune tolerance [6]. PD-L2 can also bind to repulsive guidance molecule b. Blockade of the repulsive guidance molecule b–PD-L2 interaction markedly impaired the development of respiratory tolerance by interfering with the initial T-cell expansion required for respiratory tolerance [7]. Previous studies showed that T-cell activity in PD-L2 knockout mice was enhanced, the amounts of activated CD4+/CD8+ T cells stimulated by exogenous antigen were increased, and PD-L2 knockout also resulted in the disappearance of T-cell tolerance against foreign antigens [6]. In general, the functions of PD-L1 and PD-L2 overlap, but they also have their own unique features. PD-L1 has priority in controlling the interaction of T cells and antigen-presenting cells, inhibiting PD-L1-enhanced hapten-induced contact hypersensitivity (CH) in a mouse model, and increasing the number of T cells in draining lymph nodes, whereas PD-L2 inhibition did not show a similar strengthening effect [8]. On the other hand, PD-L2 possesses the ability to drive the Th1 cell response through IFN-γ, and signaling of the PD-1/PD-L2 pathway can significantly inhibit TCR-mediated CD4+ T-cell proliferation and cytokine production [9]. Blocking PD-L2 on the DC surface enhanced the proliferative capacity of T cells and the production of cytokines such as IFN-γ and IL-10 [5]. Dual inhibition of PD-L1 and PD-L2 can show a synergistic effect [5].
Since the binding of PD-L1 and PD-L2 to PD-1 is cross competitive [10], when PD-1 inhibitors anchor the binding site and conformation of PD-1, the interaction between PD-1 and both PD-L1/PD-L2 ligands can be blocked simultaneously. Therefore, PD-1 inhibitors are able to completely antagonize the PD-1/PD-L1 and PD-1/PD-L2 axes, whereas PD-L1 inhibitors selectively block only the interaction between PD-1 and PD-L1, but retain the activity of the PD-1/PD-L2 pathway [11].
PD-L1 inhibitor: PD-1/PD-L1 & B7-1/PD-L1 axis
In addition to its interaction with PD-1, PD-L1 can also bind to B7-1. Both B7-1 and B7-2 belong to the B7 family, which can interact with CD28 to initiate a series of stimulatory signals for T-cell activation [9]. In the early stages of T-cell activation, cytotoxic T lymphocyte-associated antigen 4 (CTLA-4, another type of immune checkpoint molecule) can competitively bind with B7-1/B7-2 to inhibit CD28: B7 signaling, thereby preventing T-cell activation. B7-2 has been shown to be constantly expressed at a low level on the surface of antigen-presenting cell, and can be activated rapidly; in contrast, B7-1 only showed inducible expression after B7-2 activation [12]. Previous studies have shown that PD-L1 can also bind competitively to B7-1, and there is overlap between the binding interfaces of PD-L1: B7-1, CD28: B7-1 and CTLA-4: B7-1 [12]. Therefore, PD-L1 can inhibit T-cell activation and immune effector cytokine production, along with other inhibitory pathways such as CTLA-4. However, protein equilibrium dissociation constant (KD) data showed that the KD value of PD-L1: B7-1 (KD = ∼1.7 μM) was between that for CD28: B7-1 (KD = 4 μM) and CTLA-4: B7-1 (KD = 0.2 μM). Because the KD value is inversely related to protein affinity, the greater the KD value, the weaker the affinity. Therefore, the affinity of the PD-L1: B7-1 interaction is much lower than that of CTLA-4: B7-1 [12]. Although PD-L1 inhibitors disrupt the interaction between PD-L1 and B7-1, CTLA-4 may also compensatively disqualify this ‘immunosuppressive’ effect, leaving the immune system still at the inhibited status controlled by the checkpoint molecule. In addition, the interaction between CTLA-4: B7-1 and CD28: B7 signaling often plays an important role in the early stage of T-cell activation, which weakens, inhibits and/or abrogates the activation of naive T cells in the second lymphoid organs (such as lymph nodes), while the high expression of PD-L1 is often specifically limited to tumor cells and immune cells in the tumor microenvironment [13]. Therefore, more evidence is still required to determine whether the inhibition of PD-L1 will affect the axis of PD-L1/B7-1/CD28 and the corresponding early activation process of T cells.
Isotype of monoclonal of antibody
The binding of antibodies and antigens is very specific. Via the unique variable regions of antibodies in most cases, an antibody can specifically recognize an antigen. This one-to-one correspondence relationship enables antibody drugs to have fewer adverse events and higher efficiency. Antibodies in the human body can be divided into five types, including IgA, IgD, IgE, IgG and IgM. These five types of antibodies have their own functions and different half-lives.
When antibody engineers were faced with choosing from this array to develop therapeutic agents, practical and functional concerns led them to focus on the IgG class. Even within this class, only IgG1, IgG2 and IgG4 have been used, as various considerations have largely excluded the use of IgG3 [14]. IgG1 and IgG3 have two unique characteristics: antibody-dependent cytotoxicity (ADCC) and complement-dependent cytotoxicity (CDC). Because of these two mechanisms, once IgG1 and IgG3 bind with an antigen on target cells, the cells will be deleted. This is obviously not suitable for a PD-1 antibody, which is aimed at activating T cells rather than killing T cells. But IgG2 and IgG4 subtypes have a much lower potency to mediate ADCC than IgG1 and IgG3 [15]. Therefore, IgG4 was chosen for nivolumab rather than IgG2, which binds mainly to sugar antigens. No in vitro ADCC or complement-dependent cytotoxicity activity was observed with nivolumab in assays using PD-1-expressing activated T cells as target cells, suggesting that nivolumab is unlikely to deplete PD-1-positive cells [16]. Pembrolizumab also belongs to IgG4 subclass of human antibodies [17].
For PD-L1 inhibitors, antibody design is theoretically aimed at depleting target tumor cells via an ADCC effect, so that all PD-L1 inhibitors are chosen as the IgG1 isotype. But immune checkpoint antibodies of the IgG1 isotype, such as atezolizumab [18] and durvalumab [19] are genetically engineered to eliminate ADCC activity to protect PD-1/PD-L1-positive immune cells. Avelumab is the single exception of a therapeutic antibody, which exploits immune checkpoint inhibition and ADCC-mediated killing of tumor cells simultaneously [20].
Action site & conformation
Representative PD-1 inhibitors (i.e., nivolumab, pembrolizumab) and PD-L1 inhibitors (i.e., atezolizumab, durvalumab, avelumab) are fully human or humanized monoclonal antibodies. When the antibodies bind to the epitopes of antagonist targets, they can anchor the corresponding domains and form a stable conformation through the interaction of molecular bonds. Since the binding of a molecule to its corresponding ligand/receptor, and the subsequent conformational changes are often the starting point of the delivery of signaling, downstream signals may hardly be delivered or substantially diminished once the binding sites are competitively occupied or even surrounded by the antibodies; thereby, antibodies can suppress the physiological or pathological role of the targeted molecule [21]. Structurally, there are differences in the acting domains, antigen epitopes and conformational changes between each inhibitor/molecule (i.e., each antibody/antigen), and these differences at the molecular level can be attributed to differences in antibody affinity, binding duration and clinical utility [22–24].
PD-1 is a type I transmembrane protein, major domains of which contain an extracellular immunoglobulin variable region (IgV)-like domain, a transmembrane domain and C-terminal amino acid residues in the cytoplasmic region [25]. Among them, the cytoplasmic domain of PD-1 contains two independent phosphorylation sites, namely the immunoreceptor tyrosine inhibition motif (ITIM) and the immunoreceptor tyrosine transfer motif (ITSM). A phosphorylated PD-1 peptide, corresponding to the C-terminal (ITSM), acts as a docking site in vitro for both SHP-2 and SHP-1, while the phosphorylated peptide containing the N-terminal PD-1 (ITIM) associates only with SHP-2 [26]. When PD-1 binds to its ligand, tyrosine phosphorylation occurs in the intracellular domain. Phosphorylated ITSM can recruit SHP-1 and SHP-2, and cause the dephosphorylation of downstream signaling molecules such as PIK3/AKT, ZAP70, CD3ξ and PKCθ, and thus inhibit the antigen signal and other costimulatory signals from transferring into the nucleus [1]. Consequently, these motifs are implicated in its immunosuppressive effects. Interfering with PD-1 signal transduction, either by antibody blockade or any other means, enhances T-cell functions by potentiating signal transduction from the TCR signalosome [27]. On the other hand, as a member of the B7 family, PD-L1/PD-L2 molecules are type I transmembrane glycoproteins with an IgV and an immunoglobulin constant region (IgC)-like region in the extracellular domain that is connected to a short, charged cytoplasmic zone through a hydrophobic transmembrane region [25]. The homology among PD-L1 and PD-L2 in human is 40% [28].
Although the binding epitope is different for each inhibitor, similar inhibitor–antigen interactions can be observed between PD-1/PD-L1 and each inhibitor. The binding of PD-1 to nivolumab or pembrolizumab directly covers part of the binding sites for PD-1 and PD-L1, and thus results in conformational changes in the BC loop (residues 57-63) and the FG loop (residues 127-124) of the PD-1 extracellular region [29]. The arrangement of the BC, FG region disrupts the PD-1/PD-L1 interaction. On the other hand, the CC'FG loop on the β-sheet of the PD-L1 extracellular region is a key domain for the binding of PD-L1 to all antibodies (Figure 2). The common antigen epitopes on PD-L1 include PD-L1Y56, E58, R113, M115 and Y123, all of which are located in the CC'FG loop, providing a key intracellular interaction force for the binding of PD-1/PD-L1 [30]. In comparison, the KD value for PD-1 and PD-L1 is about 8.2 μM, which is much higher than the KD values among all antibodies and PD-1 or PD-L1 (Table 1). As has been noted earlier, the KD value is inversely proportional to the strength of protein binding affinity so, on the contrary, all the antibodies can demonstrate far greater affinity to PD-1 or PD-L1 than that between PD-1 and PD-L1 or PD-L2 (i.e., the physiological interaction between each molecule involved in the PD-1 pathway), thus showing sufficient efficacy in blocking the PD-1 signal transmission pathway [28]. It should be noted that pembrolizumab mainly binds to the C'D loop of PD-1, whereas nivolumab mainly binds to the N-terminal loop [29]. Therefore, there are almost no overlapping antigen binding epitopes, domains and areas on PD-1 for these two antibodies [29]. Competitive binding assays also showed that the binding of nivolumab and pembrolizumab with PD-1 is partially complementary, suggesting that the simultaneous administration of pembrolizumab and nivolumab may be considered for future therapy [29,31].
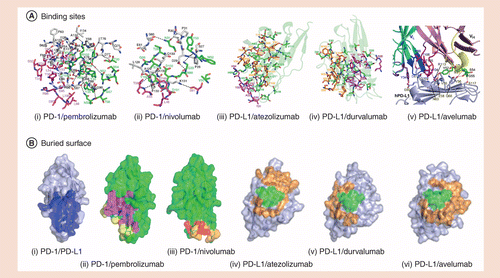
(A) Stereo view of the detailed interface of PD-1/PD-1 inhibitors (i) pembrolizumab and (ii) nivolumab. Stereo view of the detailed interface of PD-L1/PD-L1 inhibitors Fab fragment, (iii) atezolizumab, (iv) durvalumab and (v) avelumab. (B) Surface representations of the PD-1 molecules in the complex structures of PD-1/PD-L1 (i), PD-1/pembrolizumab (ii), PD-1/nivolumab (iii), PD-L1/atezolizumab (iv), PD-L1/duralumab (v) and PD-L1/avelumab (vi).
PD-1: Programmed cell death-1; PD-L1: Programmed cell death ligand-1.
Reproduced with permission from [22] © Nature Communications (CC BY 4.0) (2016); Reproduced with permission from [23] © Springer Nature (2019); Reproduced with permission from [30] © Nature (CC BY 4.0) (2017).
| Molecular characteristics | Parameters (PD-1 or PD-L1 with each inhibitor) | ||||
|---|---|---|---|---|---|
| Nivolumab | Pembrolizumab | Atezolizumab | Durvalumab | Avelumab | |
| Acting domain | N-loop; FG, BC loop | C'D loop | CC'FG antiparallel β-sheet; BC, CC’, C'C’’ and FG loops | Parallel β-sheet; A, G, F and CC’ loops/strands | Parallel β-sheet; CC'FG loop/strand |
| Major binding sites | D29, R30, S60, K131; P28, L128, A129, etc. | N66, T76, K78, S87, K131; F63, E84, S87, etc. | E58, Q66, V111 (hydrogen bond); R113, R125 (salt bridge) | D26, R113; Y123, K124, R125 (hydrogen bond) | I54, Y56, M115, Y123 (hydrogen bond); E58, Q66, R113, A121 |
| Buried surface | 1349 Å | 2126 Å | 2106 Å | 1624 Å | 1865 Å |
| Affinity (KD) | 1.45 nM | 400 pM | 1.75 nM | 0.667 nM | 0.046 nM |
Pharmacokinetic & pharmacodynamic characteristics
Taking nivolumab as an example, Phase I clinical trials showed that nivolumab had a high PD-1 occupancy, with a mean peak occupancy of 85% (range: 70–97%) [32], which was consistent with the high PD-1 affinity of nivolumab observed in in vitro studies [33]. Nivolumab at a trace dose as low as 0.04 ng/ml was sufficient to occupy >70% of PD-1 molecules on the surface of T cells, suggesting that even though the serum concentrations of nivolumab in patients’ peripheral blood were below the detection threshold (<1.2 μg/ml), the drug concentration was still sufficient to maintain relatively high PD-1 occupancy in the tumor microenvironment [33]. In addition, nivolumab exhibited durable PD-1 occupancy, with plateau occupancy being maintained even when serum concentrations were undetectable. Regardless of the administered dose, a sustained mean occupancy of >70% of PD-1 molecules on circulating T cells was observed for as long as 2 months after infusion [33].
The Checkmate 077 trial (cohort A) evaluated the pharmacokinetic profile of nivolumab in small-scale Chinese NSCLC (n = 9) and nasopharyngeal cancer (n = 6) populations [34]. The pharmacokinetic profile of nivolumab in the Chinese population was found to be similar to that in western populations. The peak plasma concentration, time to peak concentration, area under the plasma concentration time curve and half-life of nivolumab in the Chinese population showed no significant differences compared with values obtained in a western population [35]. The pharmacokinetic and pharmacodynamic parameters of some other PD-1/PD-L1 inhibitors are shown in Table 2, although specific data are not available in all cases. As shown in Table 2, pharmacokinetic/pharmacodynamic parameters determine the standard dosage, frequency and schedule of administration for each drug. In general, all of the PD-1/PD-L1 inhibitors can maintain a relatively high drug concentration and sustained or durable antitumor efficacy.
| Pharmacokinetics & pharmacodynamic characteristics | Parameters (PD-1 or PD-L1 with each inhibitor) | ||||
|---|---|---|---|---|---|
| Nivolumab | Pembrolizumab | Atezolizumab | Durvalumab | Avelumab | |
| Mean peak occupancy | 85% (70–97%) [36] | N/A | N/A | N/A | N/A |
| Median/target occupancy | 64–70% [37] | ∼60% [38] | >65% [36] | N/A | 76–93% (mean value) [39] |
| Mean plateau occupancy | 72% (59–81%) [36] | N/A | N/A | N/A | N/A |
| Time to steady state (weeks) | 12 weeks (Q2W) [36] | 19 weeks (Q3W) [36] | 6–9 weeks (Q3W) [36] | 16 weeks (Q2W) [36] | 4–6 weeks (Q2W) [36] |
| Cumulative dose | 3.7-fold [36] | 2.2-fold [36] | 1.91-fold (AUC) [36] | 4.3-fold [36] | 1.25-fold [36] |
| Mean elimination half-life (t1/2, days) | 25 [36] | 23 [36] | 27 [36] | 17 [36] | 6.1 [36] |
Clinical efficacy & safety
NSCLC is one of the most widely understood and clinically applied fields for PD-1/PD-L1 inhibitors. Currently, four PD-1/PD-L1 inhibitors, including two PD-1 inhibitors (nivolumab and pembrolizumab) and two PD-L1 inhibitors (atezolizumab and durvalumab) have been approved for the treatment of locally advanced/metastatic NSCLC. The results of representative randomized, controlled studies in NSCLC are shown in Table 3.
| Drug | Trial (Phase) | Study population | Patients (n) | ORR (%) | mPFS (months) | mOS (months) |
|---|---|---|---|---|---|---|
| PD-1 inhibitors | ||||||
| Nivolumab [40] | Checkmate 017 (III) | Previously treated advanced/metastatic squamous cell carcinoma | 272 | 20 | 3.5 | 9.2 |
| Nivolumab [41] | Checkmate 057 (III) | Previously treated advanced/metastatic nonsquamous cell carcinoma | 582 | 19.2 | 2.3 | 12.2 |
| Nonsquamous cell carcinoma subgroup; PD-L1 ≥1% | – | 30.9 | – | 17.1 | ||
| Pembrolizumab [42] | KEYNOTE-001 (I) | Previously treated advanced/metastatic NSCLC; PD-L1 ≥1% | 495 | 19.4 | 3.7 | 12.5 |
| Pembrolizumab [29] | KEYNOTE-010 (II/III) | Previously treated advanced/metastatic NSCLC; PD-L1 ≥1% | 1034 | 18 | 4.0 | 10.4 |
| PD-L1 inhibitors | ||||||
| Atezolizumab [43] | POPLAR (II) | Previously treated advanced/metastatic NSCLC | 287 | 15 | 2.8 | 11.4 |
| Atezolizumab [44] | OAK (III) | Previously treated advanced/metastatic NSCLC | 1225 | 14 | 2.8 | 13.8 |
| Nonsquamous cell carcinoma; PD-L1 ≥1% | – | 18 | – | 17.6 | ||
| Avelumab [45] | NCT01772004 (I) | Previously treated advanced/metastatic NSCLC | 184 | 12 | 2.9 | – |
Despite a series of studies focusing on comparisons of PD-1/PD-L1 inhibitors with standard chemotherapy in the subsequent line treatment setting of NSCLC, only nivolumab-related studies divided NSCLC patients into squamous cell carcinoma and nonsquamous cell carcinoma subgroups, according to the patients’ histological features, and allowed separate subanalyses to be conducted. The Checkmate 017 study, which was conducted in patients with squamous cell carcinomas (n = 272) [40], showed that the objective response rate (ORR: 20 vs 9%; p = 0.008), median overall survival (mOS: 9.2 vs 6.0 months; p < 0.001) and median progression-free survival (mPFS: 3.5 vs 2.8 months; p < 0.001) were all significantly improved in nivolumab-treated patients compared with patients receiving docetaxel. Regardless of the PD-L1 expression status, patients with advanced squamous cell carcinoma can benefit from second-line nivolumab treatment. Outcomes in the Checkmate 057 study (n = 582), which was conducted in patients with nonsquamous cell carcinoma, were similar to those observed in patients with squamous cell carcinoma [41]. The overall ORR (19 vs 12%; p = 0.008) and mOS (12.2 vs 9.4 months; p = 0.002) were significantly improved in nivolumab-treated patients in comparison with those receiving docetaxel. However, patients with positive PD-L1 expression (≥1%) had a better response to nivolumab (31 vs 12%), and the survival benefit with nivolumab compared with chemotherapy was more significant in these patients (hazard ratio = 0.59).
Participants in subsequent line studies of pembrolizumab treatment involved the overall population of NSCLC patients with positive PD-L1 expression (i.e., PD-L1 ≥1%). The KEYNOTE-001 study (n = 495) [42] demonstrated that regardless of the histological type, the overall ORR of PD-L1-positive patients who received pembrolizumab treatment was 19.4%, and the mOS was 12.0 months. The KEYNOTE-010 study, a randomized controlled study comparing pembrolizumab with docetaxel, showed that the ORR (18 vs 9%, respectively) and mOS (10.4 vs 8.5 months, respectively; p = 0.0008) were significantly improved in the pembrolizumab arm compared with the chemotherapy arm. However, no significant difference in mPFS (4.0 vs 4.0 months, respectively; p = 0.07) was observed [46]. Similar to the results obtained with nivolumab in patients with nonsquamous cell carcinoma, patients with higher PD-L1 expression (such as PD-L1 ≥50%) showed an improved response and survival benefits with pembrolizumab.
Similar to studies with pembrolizumab, participants in subsequent line studies of atezolizumab treatment involved the overall population of NSCLC patients without stratification according to the histological type. However, corresponding retrospective subanalyses were performed for subgroups of squamous or nonsquamous cell carcinoma and patients were stratified according to their PD-L1 expression status. Results from the OAK study (n = 1225) [44] showed no significant differences in ORR (14 vs 13%, respectively) and mPFS (2.8 vs 4.0 months, respectively) between the atezolizumab or docetaxel treatment arms, but mOS in atezolizumab-treated patients was significantly improved (13.8 vs 9.6 months, respectively; p = 0.0003). Regardless of whether patients were positive or negative for PD-L1 expression or had squamous or nonsquamous cell carcinoma, a survival benefit (in mOS) was observed with atezolizumab treatment [44].
Further studies of similar populations to those in the above studies may provide more information on the comparative effectiveness of each inhibitor. An intuitive, horizontal comparison of the PD-1 inhibitor nivolumab with the PD-L1 inhibitor atezolizumab in PD-L1-positive (PD-L1 ≥1%) nonsquamous cell carcinoma patients, showed a numerically higher ORR in patients treated with nivolumab than in those treated with atezolizumab (30.9 vs 18%, respectively) [41,44]. A pooled analysis of large-scale, randomized, controlled trials (n = 4755) of PD-1/PD-L1 inhibitors in NSCLC, advanced melanoma and genitourinary cancer indirectly compared the efficacies of each drug. The ORRs of nivolumab-treated patients in PD-L1-positive and -negative subgroups were 39.3% (34.1∼44.4%) and 22.9% (19.4∼26.3%, p < 0.0001), respectively; corresponding rates in patients treated with pembrolizumab were 30.3% (25.2−35.3%) and 10.8% (3.3−18.4%), respectively. Due to limited data, pooled statistical analysis for atezolizumab was not performed, but the ORR of atezolizumab-treated patients ranged from 20 to 29% and from 10 to 26% in PD-L1-positive and -negative subgroups, respectively [47]. Recently, a meta-analysis of five trials involving 3024 NSCLC patients found that indirect comparisons of immune checkpoint inhibitors revealed little evidence of differences between nivolumab, pembrolizumab and atezolizumab [48]. However, in terms of safety, a recently published paper showed that nivolumab had a narrow and mild toxicity spectrum, which makes it the best option so far, especially for the treatment of lung cancer [49].
In general, there have been no significant differences in efficacy between PD-1 and PD-L1 inhibitors in large-scale clinical trials. Definitive conclusions about comparative efficacy should therefore be based on population-based studies or large-scale head-to-head comparative studies.
To date, no head-to-head study has been performed to compare the safety of each PD-1/PD-L1 inhibitor; therefore, safety comparisons among the drugs cannot be evaluated directly, but results from large-scale Phase III clinical studies may provide some useful information [40,41,43,44,46,50]. Overall, both the incidence of any grade of adverse events (AEs) and serious AEs induced by PD-1 or PD-L1 inhibitors are significantly lower than with traditional treatment regimens such as chemotherapy. The safety profile of PD-1/PD-L1 inhibitors is distinct from that observed with chemotherapy; AEs, and mainly include fatigue, skin/gastrointestinal/kidney/liver/endocrine system-related events and toxicity, and pneumonia. When the incidence of any grade of AEs, high-grade AEs (grade 3–5), treatment-related AEs, or common symptoms such as fatigue, diarrhea and rash, is considered, no obvious numerical differences were found, suggesting that the safety characteristics among PD-1/PD-L1 inhibitors are similar (all p > 0.3; see Table 4 for details). However, population-based studies or large-scale head-to-head comparisons are still required to provide more robust clinical evidence.
| Adverse events (AEs) | PD-1 inhibitors | PD-L1 inhibitors | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Nivolumab, % [40,41,51–59] | Pembrolizumab, % [46,50,60–64] | Atezolizumab, % [43,65,66] | Durvalumab, % [67] | Avelumab, % [68,69] | ||||||
| Any grade | ≥3 grade | Any grade | ≥3 grade | Any grade | ≥3 grade | Any grade | ≥3 grade | Any grade | ≥3 grade | |
| Any AE (%) | 58–82 | 7–22 | 61–82 | 10–27 | 66–94 | 16–40 | ∼64 | ∼5 | 78 | 13 |
| Skin | ||||||||||
| – Pruritus | 7–29 | <1 | 11–21 | 1 | 11–14 | 0–1 | ∼3 | 0 | NR | NR |
| – Rash | 6–26 | <1 | 10–21 | 2 | 9–14 | 0–1 | NR | NR | 13 | 0 |
| – Vitiligo | 10–11 | <1 | 9–11 | <1 | NR | NR | NR | NR | NR | NR |
| Gastrointestinal | ||||||||||
| – Diarrhea | 7–16 | 0–2 | 8–20 | 1 | 11–20 | 0–2 | ∼10 | 0 | 7 | 0 |
| – Colitis | 1–3 | 1 | 1–4 | 1–2 | 1 | <1 | NR | NR | NR | NR |
| Liver | ||||||||||
| – Elevated ALT levels | 1–4 | <1 | 2–8 | <1 | 2–4 | 0–3 | NR | NR | 1 | 1 |
| – Elevated AST levels | 1–4 | <1 | 3–10 | 1 | 2–3 | 0–3 | NR | NR | ||
| Endocrine system | ||||||||||
| – Hypothyroidism | 4–9 | <1 | 8–10 | <1 | 7–9 | 0 | NR | NR | 3–6 | 0 |
| – Hyperthyroidism | 0–3 | <1 | 3–8 | <1 | 1 | <1 | NR | NR | 2 | 0 |
| – Hypophysitis | <1 | <1 | <1 | <1 | <1 | <1 | NR | NR | NR | NR |
| Others | ||||||||||
| – Pneumonia | 1–5 | 0–2 | 4–6 | 1–2 | 3 | <1 | NR | NR | 1 | 0–1 |
| – Nephritis/acute kidney injury | 1–3 | 0–2 | <1 | <1 | 2 | 2 | ∼2 | ∼2 | NR | NR |
| – Neurotoxicity | <1 | <1 | <1 | <1 | NR | NR | NR | NR | NR | NR |
| Treatment stopped due to irAE | 5–8 | 4–7 | 4–8 | ∼2 | 2 | |||||
When monotherapy with PD-1/PD-L1 drugs in advanced MM is considered (Table 5), two PD-1 blockers, nivolumab and pembrolizumab have been tested in Phase III trials and approved for the treatment of nonresectable and metastatic MM. The approval of nivolumab by the EMA was based on two studies: the CheckMate-066 and -037 studies. CheckMate-066 was a randomized, double-blind, Phase III study involving 418 previously untreated BRAF wild-type patients with unresectable or metastatic MM. The primary end point (overall survival [OS]) was reached in patients treated with nivolumab, and the median progression-free survival (PFS) was 5.1 months with nivolumab as compared with 2.2 months with the comparator treatment dacarbazine (HR: 0.43; 95% CI: 0.34–0.56; p < 0.001). The ORR was 40.0% with nivolumab and 13.9% dacarbazine (odds ratio: 4.06; p < 0.001). The 11.7% of AEs were grade 3 or 4, and no therapy-related deaths were reported [51]. The CheckMate-037 study of nivolumab versus chemotherapy demonstrated that patients previously treated with ipilimumab (or ipilimumab and a BRAF inhibitor in BRAFV600-positive patients) can still respond to nivolumab as compared with dacarbazine or carboplatin and paclitaxel therapy; the ORR in the two groups was 31.7 and 10.6%, respectively [70]. AEs were comparable to those in the Check-Mate 066 study. In the Phase III trial Keynote-006 study, significantly higher PFS and OS were found with pembrolizumab compared with ipilimumab (ORR was 33% in both pembrolizumab arms and 11.9% in the ipilimumab arm). However, monotherapy data with PD-L1 inhibitors are currently very limited. As summarized in Table 5, atezolizumab has demonstrated preliminary efficacy in advanced melanoma with an ORR of 26% and 24-week PFS of 41% [71].
| Drug | Phase | Study population | Treatment arm | Efficacy data | Grade 3–4 or 3–5 AEs | Ref. |
|---|---|---|---|---|---|---|
| PD-1 inhibitors | ||||||
| Nivolumab (Checkmate 066) | III (n = 418) | Unresectable stage III or IV melanoma; no prior systemic treatment; no BRAF mutation | Nivolumab vs dacarbazine | Median PFS: 5.1 vs 2.2 months; ORR: 40 vs 13.9% | 11.7 vs 17.6% | [51] |
| Nivolumab (CheckMate 037) | III (n = 405) | Unresectable stage III or IV melanoma after ipilimumab or after ipilimumab and BRAFi for BRAF mutants | Nivolumab vs investigator's choice of chemotherapy (dacarbazine or paclitaxel + carboplatin) | ORR: 31.7 vs 10.6% | 5 vs 9% | [70] |
| Pembrolizumab (KEYNOTE 006) | III (n = 834) | Unresectable stage III or IV melanoma; no more than one prior systemic treatment | Pembrolizumab (q14 or q21) vs ipilimumab | ORR: 33.7/32.9% vs 11.9% | 13.3/10.1% vs 19.9% (grade 3–5) | [51] |
| PD-L1 inhibitors | ||||||
| Atezolizumab | I (n = 43) | Advanced melanoma | Dose escalation | ORR: 26%; 24-week PFS: 41% | NA | [71] |
Thus, it is difficult to compare PD-1 and PD-L1 drugs in advanced melanoma due to the limited data available with PD-L1 therapy. A combination strategy involving a PD-1 blocker and a CTL-A4 inhibitor has recently been explored [72].
Conclusion
In recent years, the crystal structures of PD-1 and PD-L1 have been revealed, but there is continued debate concerning the implications of theoretical differences between the PD-1 and PD-L1 inhibitors in terms of clinical benefit. This review has illustrated the complicated signaling pathways of PD-1: PD-L1/PD-L2, PD-L1/CD28/CTLA-4: B7-1 axes in the immune checkpoint system. The binding affinity of each antibody to either PD-1 or PD-L1 is much higher than that between PD-1: PD-L1, with each antibody partially or completely covering the binding sites, anchoring the key domains to prevent the binding of PD-1 and PD-L1, and thus showing sufficient potency to block the negative signal of the checkpoint pathway. Generally speaking, the mechanisms of action of PD-1 and PD-L1 inhibitors are, to some extent, different with different binding targets, but they also reflect corresponding cross connectivity and each inhibitor's unique characteristics.
On the basis of current clinical data, the efficacy and safety profiles of PD-1 and PD-L1 inhibitors are promising, showing no significant differences between the two classes of drugs. However, even in horizontal comparisons of large-scale, randomized clinical trials, differences in patient characteristics and disease characteristics may still lead to bias in terms of the superiority–inferiority conclusion.
Future perspective
Many clinical trials of PD-1/PD-L1 inhibitors have been conducted in different solid tumors. While some PD-1 drugs have achieved positive results, some PD-L1 drugs have produced negative results in the same tumor, and vice versa. Is this by chance or is there some scientific rational behind this phenomenon? Will there be any clinical differences between PD-1/PD-L1 drugs for certain tumors in the future? Perhaps only head-to-head comparative studies between active drugs can provide definitive information regarding efficacy and safety differences between specific PD-1/PD-L1 drugs. In addition, there remains an unresolved issue as to whether PD-L1 drugs could be used as subsequent lines of therapy after resistance has occurred to PD-1 inhibitor treatment, and vice versa. Recently, some studies have been initiated to investigate this issue. Thus, our review is quite critical in highlighting important future considerations, not only in terms of clinical treatment decisions for specific tumor types, but also possibly in terms of changes in immunotherapy treatment.
Executive summary
Background
Programmed cell death-1 (PD-1)/programmed death-ligand 1 (PD-L1) checkpoint inhibitors are used widely in many types of solid tumors, and are often considered to be in the same immunotherapy subclass.
Due to the complicated functional pathways in the immune checkpoint system, the epitopes, interface and signal pathways between PD-1: PD-L1/PD-L2, PD-L1/CD28/CTLA-4:B7-1, axes often overlap and affect each other.
Therefore, the mechanisms of action of PD-1 and PD-L1 inhibitors reflect corresponding cross connectivity and unique characteristics of each agent.
Binding affinities
The binding affinity of each antibody to either PD-1 or PD-L1 is much higher than that between PD-1: PD-L1, with each antibody partially or completely covering the binding sites, anchoring the key domains to prevent the binding of PD-1 and PD-L1.
PD-1 and PD-L1 inhibitors therefore exhibit sufficient potency to block the negative checkpoint pathway signal.
Clinical efficacy & safety
On the basis of current clinical data, the efficacy and safety profiles of PD-1 and PD-L1 inhibitors are promising, showing no significant differences between the two classes of drugs.
Many clinical trials of PD-1/PD-L1 inhibitors have been conducted in different solid tumors. While some PD-1 drugs have achieved positive results, some PD-L1 drugs have had negative results in the same tumor, and vice versa.
In general, there has been no significant difference in the efficacy between PD-1 and PD-L1 inhibitors in large-scale clinical trials.
Whether PD-L1 drugs could be used as subsequent lines of treatment after resistance has occurred to PD-1 inhibitor treatment, and vice versa, remains an unresolved issue.
Only head-to-head comparative studies can provide definitive information regarding clinical efficacy and safety differences between specific PD-1/PD-L1 inhibitors.
Financial & competing interests disclosure
This review was supported by National Natural Science Foundation of China (Grant No. 81672984), National Key Research and Development Plan (Grant No. 2017YFC0907900). The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
Medical writing support was provided by DP Figgitt, ISMPP CMPP™ and editorial assistance was provided by Content Ed Net in the preparation of this manuscript, with funding from BMS China.
Open access
This work is licensed under the Attribution-NonCommercial-NoDerivatives 4.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
Papers of special note have been highlighted as: • of interest
References
- 1 . The PD1:PD-L1/2 pathway from discovery to clinical implementation. Front. Immunol. 7, 550 (2016). • Overview on the properties of the components of the programmed cell death-1 (PD-1) pathway, the signaling events regulated by PD-1 engagement and their consequences on the function of T-effector cells.
- 2 PD-1 and PD-L1 checkpoint signaling inhibition for cancer immunotherapy: mechanism, combinations, and clinical outcome. Front. Pharmacol. 8, 561 (2017).
- 3 PD-L2 expression in human tumors: relevance to anti-PD-1 therapy in cancer. Clin. Cancer Res. 23, 3158–3167 (2017).
- 4 Clinicopathologic analysis of PD-L1 and PD-L2 expression in renal cell carcinoma: association with oncogenic proteins status. Ann. Surg. Oncol. 23, 694–702 (2016).
- 5 Blockade of programmed death-1 ligands on dendritic cells enhances T cell activation and cytokine production. J. Immunol. 170, 1257–1266 (2003).
- 6 Regulation of T cell activation and tolerance by PDL2. Proc. Natl Acad. Sci. USA 103, 11695–11700 (2006).
- 7 RGMb is a novel binding partner for PD-L2 and its engagement with PD-L2 promotes respiratory tolerance. J. Exp. Med. 211, 943–959 (2014).
- 8 Preferential contribution of B7-H1 to programmed death-1-mediated regulation of hapten-specific allergic inflammatory responses. Eur. J. Immunol. 33, 2773–2782 (2003).
- 9 Lessons learned from the blockade of immune checkpoints in cancer immunotherapy. J. Hematol. Oncol. 11, 31 (2018).
- 10 . Human cancer immunotherapy with PD-1/PD-L1 blockade. Biomark. Cancer 7(Suppl 2), 15–18 (2015). • Review of the progress of PD-1 axis blockade in clinical trials to evaluate its effectiveness in the antitumor immunotherapy.
- 11 . PD-1 pathway inhibitors: immuno-oncology agents for restoring antitumor immune responses. Pharmacotherapy 36, 317–334 (2016). • Provides with a basic understanding of the role of PD-1 in modulating the immune system and their use in the cancer treatment.
- 12 Programmed death-1 ligand 1 interacts specifically with the B7-1 costimulatory molecule to inhibit T cell responses. Immunity 27, 111–122 (2007). • Review illustrates a substantial bidirectional inhibitory interaction between B7-1 and PD-L1, and adds an additional dimension to immunoregulatory functions of the B7:CD28 family.
- 13 . CTLA-4 and PD-1 pathways: similarities, differences, and implications of their inhibition. Am. J. Clin. Oncol. 39, 98–106 (2016).
- 14 . Isotype selection in antibody engineering. Nat. Biotech. 25, 1369–1372 (2007).
- 15 IgG4 characteristics and functions in cancer immunity. Curr. Allergy Asthma Rep. 16, 7 (2016).
- 16 In vitro characterization of the anti-PD-1 antibody nivolumab, BMS-936558, and in vivo toxicology in non-human primates. Cancer Immunol. Res. 2, 846–856 (2014).
- 17 Structure of full-length human anti-PD1 therapeutic IgG4 antibody pembrolizumab. Nat. Struct. Mol. Biol. 22, 953–958 (2015).
- 18 Antibody-dependent cellular cytotoxicity activity of a novel anti-PD-L1 antibody avelumab (MSB0010718C) on human tumor cells. Cancer Immunol. Res. 3, 1148–1157 (2015).
- 19 . PD-L1 blockade for cancer treatment: MEDI4736. Semin. Oncol. 42, 474–483 (2015).
- 20 . Avelumab: combining immune checkpoint inhibition and antibody-dependent cytotoxicity. Expert Opin. Biol. Ther. 17, 515–523 (2017).
- 21 Mechanism of action of monoclonal antibodies that block the light activation of the guanyl nucleotide-binding protein, transducin. J. Biol. Chem. 262, 10831–10838 (1987).
- 22 Structural basis of checkpoint blockade by monoclonal antibodies in cancer immunotherapy. Nat. Commun. 7, 13354 (2016). • Review of precise epitopes of the antibodies and the molecular mechanisms underlying checkpoint blockade.
- 23 Structural basis of anti-PD-L1 monoclonal antibody avelumab for tumor therapy. Cell Res. 27, 151–153 (2017).
- 24 Distinct PD-L1 binding characteristics of therapeutic monoclonal antibody durvalumab. Protein Cell 9, 135–139 (2018).
- 25 . PD-1 and PD-1 ligands: from discovery to clinical application. Int. Immunol. 19, 813–824 (2007).
- 26 PD-1 inhibits T-cell receptor induced phosphorylation of the ZAP70/CD3zeta signalosome and downstream signaling to PKCtheta. FEBS Lett. 574, 37–41 (2004).
- 27 PD1 signal transduction pathways in T cells. Oncotarget 8, 51936–51945 (2017).
- 28 Crystal clear: visualizing the intervention mechanism of the PD-1/PD-L1 interaction by two cancer therapeutic monoclonal antibodies. Protein Cell 7, 866–877 (2016). • Focuses on the recent breakthrough work concerning the structural basis of the PD-1/programmed cell death ligands (PD-Ls) interaction and how therapeutic antibodies compete with the binding of PD-1/PD-L1 to interrupt the PD-1/PD-L1 interaction.
- 29 An unexpected N-terminal loop in PD-1 dominates binding by nivolumab. Nat. Commun. 8, 14369 (2017).
- 30 Molecular mechanism of PD-1/PD-L1 blockade via anti-PD-L1 antibodies atezolizumab and durvalumab. Sci. Rep. 7, 5532 (2017).
- 31 . Trends and advances in tumor immunology and lung cancer immunotherapy. J. Exp. Clin. Cancer Res. 35, 157 (2016).
- 32 New targeted treatments for non-small-cell lung cancer – role of nivolumab. Biologics 10, 103–117 (2016).
- 33 Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N. Engl. J. Med. 366, 2443–2454 (2012).
- 34 . Preliminary safety and pharmacokinetic results from a Phase I/II study of nivolumab in previously treated Chinese patients with advanced or recurrent solid tumors (CheckMate 077 Cohort A). Presented at: The 20th Annual Meeting of the Chinese Society of Clinical Oncology. Xiamen, China, 6–30 September (2017).
- 35 Phase I study of nivolumab, an anti-PD-1 antibody, in patients with malignant solid tumors. Invest. New Drugs 35, 207–216 (2017).
- 36 Clinical pharmacology considerations for the development of immune checkpoint inhibitors. J. Clin. Pharmacol. 57, S26–S42 (2017). • Illustrates the comprehensive investigation of complex pharmacokinetic and pharmacodynamic characteristics of PD-1/PD-L1 inhibitors as well as the definition of patient selection biomarkers.
- 37 Challenges of Phase I clinical trials evaluating immune checkpoint-targeted antibodies. Ann. Oncol. 27, 214–224 (2016).
- 38 Translational pharmacokinetic/pharmacodynamic modeling of tumor growth inhibition supports dose-range selection of the anti-PD-1 antibody pembrolizumab. CPT Pharmacometrics Syst. Pharmacol. 6, 11–20 (2017).
- 39 Avelumab for metastatic or locally advanced previously treated solid tumours (JAVELIN solid tumor): a Phase Ia, multicohort, dose-escalation trial. Lancet Oncol. 18, 587–598 (2017).
- 40 Nivolumab versus docetaxel in advanced squamous-cell non-small-cell lung cancer. N. Engl. J. Med. 373, 123–135 (2015).
- 41 Nivolumab versus docetaxel in advanced nonsquamous non-small-cell lung cancer. N. Engl. J. Med. 373, 1627–1639 (2015).
- 42 Pembrolizumab for the treatment of non-small-cell lung cancer. N. Engl. J. Med. 372, 2018–2028 (2015).
- 43 Atezolizumab versus docetaxel for patients with previously treated non-small-cell lung cancer (POPLAR): a multicentre, open-label, Phase II randomised controlled trial. Lancet 387, 1837–1846 (2016).
- 44 Atezolizumab versus docetaxel in patients with previously treated non-small-cell lung cancer (OAK): a Phase III, open-label, multicentre randomised controlled trial. Lancet 389, 255–265 (2017).
- 45 . PD-1/PD-L1 immune checkpoint blockade in non-small-cell lung cancer. Clin. Adv. Hematol. Oncol. 13, 676–683 (2015).
- 46 Pembrolizumab versus docetaxel for previously treated, PD-L1-positive, advanced non-small-cell lung cancer (KEYNOTE-010): a randomised controlled trial. Lancet 387, 1540–1550 (2016).
- 47 Differential activity of nivolumab, pembrolizumab and MPDL3280A according to the tumor expression of programmed death-ligand-1 (PD-L1): sensitivity analysis of trials in melanoma, lung and genitourinary cancers. PLoS ONE 10, e0130142 (2015).
- 48 . Comparative effectiveness of immune-checkpoint inhibitors for previously treated advanced non-small-cell lung cancer – a systematic review and network meta-analysis of 3024 participants. Lung Cancer 115, 84–88 (2018). • A comprehensive meta-analysis showing dose–response relationship between PD-L1 expression and survival benefits, and little evidence of survival differences between nivolumab, pembrolizumab and atezolizumab.
- 49 Comparative safety of immune checkpoint inhibitors in cancer: systematic review and network meta-analysis. BMJ 363, k4226 (2018).
- 50 Pembrolizumab versus chemotherapy for PD-L1-positive non-small-cell lung cancer. N. Engl. J. Med. 375, 1823–1833 (2016).
- 51 Nivolumab in previously untreated melanoma without BRAF mutation. N. Engl. J. Med. 372, 320–330 (2015).
- 52 Nivolumab for recurrent squamous-cell carcinoma of the head and neck. N. Engl. J. Med. 375, 1856–1867 (2016).
- 53 Nivolumab in metastatic urothelial carcinoma after platinum therapy (CheckMate 275): a multicentre, single-arm, Phase II trial. Lancet Oncol. 18, 312–322 (2017).
- 54 Nivolumab monotherapy in recurrent metastatic urothelial carcinoma (CheckMate 032): a multicentre, open-label, two-stage, multi-arm, Phase I/II trial. Lancet Oncol. 17, 1590–1598 (2016).
- 55 Nivolumab for classical Hodgkin's lymphoma after failure of both autologous stem-cell transplantation and brentuximab vedotin: a multicentre, multicohort, single-arm Phase II trial. Lancet Oncol. 17, 1283–1294 (2016).
- 56 Nivolumab monotherapy for first-line treatment of advanced non-small-cell lung cancer. J. Clin. Oncol. 34, 2980–2987 (2016).
- 57 Nivolumab alone and nivolumab plus ipilimumab in recurrent small-cell lung cancer (CheckMate 032): a multicentre, open-label, Phase I/II trial. Lancet Oncol. 17, 883–895 (2016).
- 58 Nivolumab versus everolimus in advanced renal-cell carcinoma. N. Engl. J. Med. 373, 1803–1813 (2015).
- 59 Nivolumab treatment for oesophageal squamous-cell carcinoma: an open-label, multicentre, Phase II trial. Lancet Oncol. 18, 631–639 (2017).
- 60 Pembrolizumab versus ipilimumab in advanced melanoma. N. Engl. J. Med. 372, 2521–2532 (2015).
- 61 Pembrolizumab versus investigator-choice chemotherapy for ipilimumab-refractory melanoma (KEYNOTE-002): a randomised, controlled, Phase II trial. Lancet Oncol. 16, 908–918 (2015).
- 62 Phase Ib study of pembrolizumab (MK-3475; anti-PD-1 monoclonal antibody) in Japanese patients with advanced melanoma (KEYNOTE-041). Cancer Chemother. Pharmacol. 79, 651–660 (2017).
- 63 Pembrolizumab as second-line therapy for advanced urothelial carcinoma. N. Engl. J. Med. 376, 1015–1026 (2017).
- 64 Pembrolizumab for platinum- and cetuximab-refractory head and neck cancer: results from a single-arm, Phase II study. J. Clin. Oncol. 35, 1542–1549 (2017).
- 65 Atezolizumab, an anti-programmed death-ligand 1 antibody, in metastatic renal cell carcinoma: long-term safety, clinical activity, and immune correlates from a Phase Ia study. J. Clin. Oncol. 34, 833–842 (2017).
- 66 Atezolizumab as first-line treatment in cisplatin-ineligible patients with locally advanced and metastatic urothelial carcinoma: a single-arm, multicentre, Phase II trial. Lancet 389, 67–76 (2017).
- 67 Safety and efficacy of durvalumab (MEDI4736), an anti-programmed cell death ligand-1 immune checkpoint inhibitor, in patients with advanced urothelial bladder cancer. J. Clin. Oncol. 34, 3119–3125 (2016).
- 68 Avelumab in patients with chemotherapy-refractory metastatic Merkel cell carcinoma: a multicentre, single-group, open-label, Phase II trial. Lancet Oncol. 17, 1374–1385 (2016).
- 69 Avelumab for patients with previously treated metastatic or recurrent non-small-cell lung cancer (JAVELIN solid tumor): dose-expansion cohort of a multicentre, open-label, Phase Ib trial. Lancet Oncol. 18, 599–610 (2017).
- 70 Nivolumab versus chemotherapy in patients with advanced melanoma who progressed after anti-CTLA-4 treatment (CheckMate 037): a randomised, controlled, open-label, Phase III trial. Lancet Oncol. 16, 375–384 (2015).
- 71 Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature 515, 563–567 (2014).
- 72 . Immunotherapy of patients with metastatic melanoma. Chin. Clin. Oncol. 6, 20 (2017).

